Regular Articles
Discovery of Novel PRMT5 Inhibitors by Virtual Screening and Biological Evaluations
2019 年 67 巻 4 号 p. 382-388
詳細
2019 年 67 巻 4 号 p. 382-388
As an important epigenetics related enzyme, protein arginine methyltransferase 5 (PRMT5) has been confirmed as an anticancer therapeutic target in recent years. Among all the reported PRMT5 inhibitors, two small molecules (GSK-3326595 and JNJ-64619178) are currently being assessed in clinical trial. In this study, 40 PRMT5 inhibitor candidates were purchased from SPECS database supplier according to the pharmacophore and molecular docking based virtual screening results. Alpha linked immunosorbent assay (LISA) methylation assay was performed to test their inhibitory activity against PRMT5. The in vitro enzymatic assay results indicated that four compounds (2, 4, 10 and 37) showed PRMT5 inhibitory activity, while 4 and 10 displayed the most potent activity with IC50 values of 8.1 ± 1.1 and 6.5 ± 0.6 µM, respectively. The inhibitory activity results of 20 extra analogs of 4 further confirmed the potency of this scaffold. As expected, compounds 4 and 10 exhibited moderate anti-proliferative activity against mantle cell lymphoma Jeko-1 and leukemia cell MV4-11. Besides, Western blot assay results showed that 4 could reduce the H4R3me2s level in a dose-dependent manner, indicating that it could inhibit the activity of PRMT5 in cellular context. Detailed interactions between 4 and PRMT5 were characterized by binding mode analysis through molecular docking. The compounds discovered in this study will inspire medicinal chemists to further explore this series of PRMT5 inhibitors.
Protein arginine methyltransferases (PRMTs) are post-translational modification related enzymes that have been confirmed to play key roles in diverse biological processes, such as cell growth and proliferation, apoptosis, metastasis, etc.1) All the biological functions of PRMTs are achieved by catalyzing the methylation of their substrate arginine residues. The identified nine PRMTs family members (PRMT1–9) can be grouped into type I (PRMT1–4, -6 and -8), type II (PRMT5 and PRMT9), and type III (PRMT7), according to the states (asymmetric dimethylarginine, symmetric dimethylarginine, and monomethylarginine) of their substrate arginine methylation levels. Among all the PRMT members, PRMT5 has recently become a hot topic of increasing interest in pharmaceutical research as a promising anticancer drug target. PRMT5 pertains to the type II PRMTs that could symmetrically transfer two methyls from S-adenosyl methionine (SAM) to its substrate proteins on arginine residues sidechains guanidine nitrogen atoms.2) While the detailed mechanisms of PRMT5 regulating the transcription, RNA metabolism, cellular differentiation and signal transduction have not been fully elucidated, increasing evidences suggest that PRMT5 is a promising anticancer target.3–14) In addition, in lymphoma, glioma and multiple myeloma (MM), PRMT5 has already been validated to be a druggable target.6,9,15)
As a consequence, mounting devotions have been taken to develop small-molecule PRMT5 inhibitors.9,16–27) The formerly reported PRMT5 inhibitors are summarized in Fig. 1. Except GSK-3326595 and JNJ-64619178 that are being evaluated in clinical trials, most of them are lack of druggability. Considering the promising anticancer target role and the limited known PRMT5 inhibitor scaffolds, development of PRMT5 inhibitors with novel skeletons is still particularly needed. Virtual screening methods, especially molecular docking, has become a very popular approach that has been successfully used to identify modulators of diverse key targets of human diseases.16,27–35) Our group has reported several PRMT5 inhibitors that were identified by applying virtual screening methods.16,27,35)

In the present study, we described the identification of four novel PRMT5 inhibitors, of which compounds 4 and 10 exhibited moderate anti-proliferative effects on mantle cell lymphoma Jeko-1 and leukemia cell MV4-11. In addition, Western blot assay results showed that 4 could reduce the H4R3me2s level in a dose-dependent manner, which suggested that 4 could target PRMT5 and inhibit its activity in cells. The binding mode of 4 with PRMT5 was proposed by using molecular docking results. The scaffold of PRMT5 inhibitor reported here provided a candidate for further structure modification to obtain more potent lead compounds.
As shown in Fig. 2a, a total of eight features (two hydrogen bond acceptors, two hydrogen bond donors, two hydrophobics, one positive ionizable and one ring_aromatic) match the PRMT5-EPZ015666 (PDB code: 4X61) interactions, and five pharmacophores were generated (Table 1). The five pharmacophores were then respectively used to screen SPECS database that contains 211838 compounds, and 1136 candidates that matched four, five and six features of each pharmacophore were retrieved. Subsequently, the 1136 candidates were docked into substrate binding site on PRMT5 by molecular docking simulation to further study their binding affinities with PRMT5. The top 300 compounds ranked by the XP Gscore were obtained. Finally, according to the clustering results, 40 PRMT5 inhibitor candidates were selected and purchased from SPECS database supplier for the inhibitory activity test against PRMT5. The docking scores and ranks of the 40 candidates were shown in Supplementary Materials (SM, Table S1), and the principal components analysis (PCA) was performed to describe the chemical space diversity of the top 300 compounds (Fig. S1, SM). The workflow of the virtual screening method employed in this study was shown in Fig. 2b.

(a) The eight pharmacophore features generated by Discovery Studio 3.0. (b) Workflow of the virtual screening method employed in this study.
| Pharmacophore | Number of features | Feature set | Selectivity score |
|---|---|---|---|
| Pharmacophore_01 | 6 | ADDHHR | 4.0549 |
| Pharmacophore_02 | 6 | ADDHPR | 4.0549 |
| Pharmacophore_03 | 6 | AADHPR | 3.6562 |
| Pharmacophore_04 | 6 | AADHHR | 3.6562 |
| Pharmacophore_05 | 6 | AADHHP | 3.0911 |
‘A’ represents hydrogen bond acceptor, ‘D’ represents hydrogen bond donor, ‘H’ represents hydrophobic, ‘R’ represents ring_aromatic, and ‘P’ represents positive ionizable.
The inhibitory activity evaluation of our purchased compounds was performed by the method that we have previously reported.16,27) Firstly, the inhibitory activity of these compounds against PRMT5 at the concentration of 50 µM was tested, and then the IC50 values of those with inhibition ratio above 50% were determined. The results showed that compounds 2, 4, 10, and 37 exhibited PRMT5 inhibitory activity, of which 4 and 10 displayed the best activity with IC50 values of 8.1 ± 1.1 and 6.5 ± 0.6 µM, respectively. Given the potency, molecular size and analog numbers in SPECS database, more analogs of 4 were searched and purchased to test their PRMT5 inhibitory activity so as to perform a structure–activity relationship (SAR) analysis. As shown in Table 2, though no more potent hits were identified among the 20 additional derivatives, the SAR provided lots of clues for further structure modification.
 |
‘NT’ represents not tested.
As compound 4 was the most active one among this class of molecules, its probable binding mode with PRMT5 was analyzed. As shown in Fig. 3a, 4 displayed a slightly different space orientations with EPZ015666 and located in the hydrophobic pocket that is composed of residues P311, L312, P314, L319, Y324, F327, P501, V503, I582, Y633 and I635. Hydrophobic, salt bridge, hydrogen bond, and π–π stacking were found to contribute to the binding affinity between 4 and PRMT5 (Fig. 3b).

(a) The panoramic and the close-up view of the binding mode of 4. For comparison, SAM and EPZ015666 were also shown, and SAM, 4 and EPZ015666 were shown as sticks while PRMT5 was shown as cartoon. (b) The interactions between 4 and PRMT5.
Two different cell lines (Jeko-1, mantle cell lymphoma cell line and MV4-11, leukemia cell line) that were previously documented to validate the anti-proliferative effects of PRMT5 inhibitors9,16) were used in this study to test the anti-proliferative activity of 4 and 10. As shown in Figs. 4a, b, 4 and 10 exhibited a dose-dependent action in inhibiting Jeko-1 and MV4-11 cell proliferation with 4-d IC50 values of 26.13, 58.35, 32.90, and 10.43 µM, respectively.
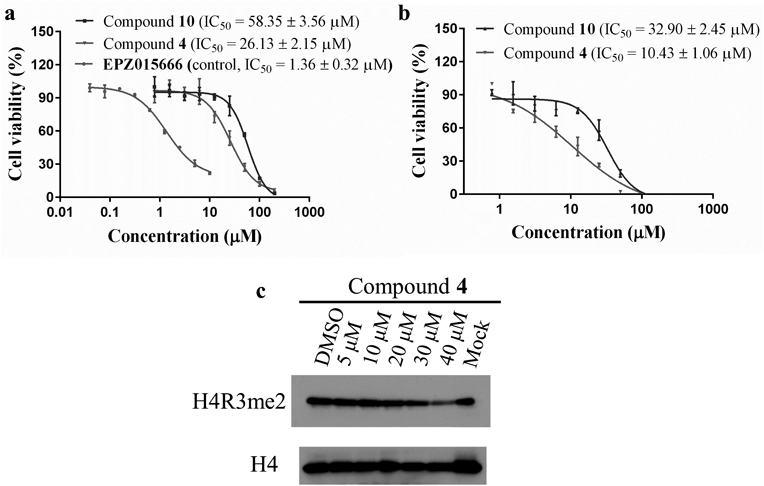
(a) The dose-inhibition curves of 4 in Jeko-1 cells. Results shown are mean ± standard deviation (S.D.) of three independent assays. (b) The dose-inhibition curves of 4 in MV4-11 cells. Results shown are mean ± S.D. of three independent assays. (c) Symmetric arginine dimethylation alterations of H4R3 in Jeko-1 cells after treatment with 4.
To further confirm the target engagement of compound 4 in cells, the effects of 4 on cellular symmetric arginine dimethylation in Jeko-1 cells were measured by immunoblot using symmetric dimethyl arginine (H4R3me2s) antibody of H3R4. As shown in Fig. 4c, treatment of 4 resulted in a concentration dependent decrease in the intensity of symmetric arginine dimethylation of PRMT5 substrate H4R3, which demonstrated that 4 could target PRMT5 and inhibit its activity at cellular level.
Here, four new PRMT5 inhibitors with novel scaffolds were discovered by structure-based virtual screening, of which compounds 4 and 10 displayed the most potent activity with IC50 values of 8.1 ± 1.1 and 6.5 ± 0.6 µM, respectively. Twenty analogs of 4 were also purchased and tested for their PRMT5 inhibitory activity. Although all of them showed weaker activity than 4, the results further confirmed the potency of this scaffold. At cellular level, compounds 4 and 10 exhibited moderate anti-proliferative activity against Jeko-1 and MV4-11 cell lines. In addition, 4 was evidenced to inhibit PRMT5 activity in Jeko-1 cells by Western blot assay results, which indicated that 4 could reduce the H4R3me2s level in a dose-dependent manner. Affinity profiling analysis indicated that 4 could well bind to the substrate pocket of PRMT5. The inhibitor identified in this work will provide a novel scaffold for PRMT5 inhibitor development and encourage medicinal chemists to further explore this series of PRMT5 inhibitors.
The crystal structure of PRMT5:MEP50 complexed with its inhibitor EPZ015666 (PDB code: 4X61) was used to perform pharmacophore and molecular docking based virtual screening. Firstly, the PRMT5-EPZ015666 complex structure was imported in Discovery Studio 3.0, and then the pharmacophore was created automatically. Subsequently, the pre-built 3D database was searched with the already created pharmacophore by Search DB tool of Discovery Studio 3.0. Glide 7.5 was employed to perform molecular docking based virtual screening. After the coordinates of protein were prepared by the Protein Preparation Wizard Workflow, the docking grid (centered on EPZ015666) was generated by the Receptor Grid Generation panel. Extra precision (XP) mode of Glide was adopted to do docking simulation. The top 300 docking poses ranked by XP G-score were selected for further cluster analysis and final selection. Pipeline Pilot 7.5 software was used to finish the cluster analysis. The clustering algorithm is a partitioning method in which the original data set is partitioned into ever-smaller regions that define the clusters, and a number of representative objects are chosen from the data set. The corresponding clusters are found by assigning each remaining object to each representative object, selecting the object that is the closest. The representative objects are called the cluster centers, while the other objects are the cluster members.
PRMT5 Inhibitory AssayEffects of the purchased compounds on methyltransferase activity of PRMT5:MEP50 were evaluated by using Alpha linked immunosorbent assay (LISA) as we previously described.16,27)
In Vitro Proliferative Effects AssayRPMI1640 medium supplemented with 10% fetal bovine serum (per ML medium), and 1% penicillin/streptomycin (per ML medium) was used to culture Jeko-1 and MV4-11 cells. Cell culture conditions were 37°C under 5% CO2. Then the treated (4 or dimethyl sulfoxide (DMSO)) cells were plated into 24-well plate with the density of 1 × 105 cells per well for growing four days. Subsequently, viable cell number was determined through the Cell Titer-Glo Luminescent Cell Viability assays (Promega) as we previously reported,16) and then translated into cell viability. Graphpad Prism 5.0 was employed to fit the IC50 values from the cell viability vs. concentration curves that were determined from at least three concentration-independence assays.
Western BlottingJeko-1 cells were incubated with 4 or DMSO (control) with different concentrations for four days. Then cells were lysed in 100 µL of total lysis buffer. After 5 min incubation at room temperature (r.t.), sodium dodecyl sulfate (SDS) was added to the cell lysates. Total cell lysates were resolved in 4–12% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred in for 1.5 h (80 V) onto polyvinylidene difluoride (PVDF) membrane (Millipore). Afterward, blots were blocked for 1 h in blocking buffer (5% nonfat milk in 0.1% Tween 20 phosphate buffered saline (PBS)) and incubated with primary antibodies (anti-H4R3me2s (symmetric), Active Motif, # 61187, anti-H4, Biorbyt, # orb327330-100 µL) in blocking buffer overnight at 4°C. The next day, after being washed five times with PBST (0.1% Tween 20 PBS), the blots were incubated with secondary antibody (HRP conjugated) for 1 h at r.t. Finally, the bands are read on the ChemiScope3400 imaging system.
ChemistryThe purity of purchased compounds was above 95% as provided by the SPECS database supplier.
This research work was financially supported by the National Natural Science Foundation of China (No. 81803438), Shandong Provincial Natural Science Foundation (Nos. JQ201721, ZR2017BH038), the Young Taishan Scholars Program (No. tsqn20161037), and Shandong Talents Team Cultivation Plan of University Preponderant Discipline (No. 10027).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.