Regular Articles
An Efficient and Scalable Synthesis of Quinolone 006
2019 年 67 巻 5 号 p. 481-486
詳細
2019 年 67 巻 5 号 p. 481-486
Quinolone 006 is under development as an anti-methicillin-resistant Staphylococcus aureus quinolone antibiotic. A linear synthetic route was utilized to prepare the compound on a multi-kilogram scale with an overall yield of 71%. The process was optimized by controlling the temperature and the vacuum pressure. Examples of parameters examined in an effort to control the polymorphism of the 006 active pharmaceutical ingredient are described.
Quinolones are among the most important synthetic antibacterial drugs, and have been extensively used in the clinic because of their superior pharmacokinetic, pharmacodynamic and absorption, distribution, metabolism and excretion (ADME) properties. Moreover, because many quinolone candidates have been tested in the clinic, the effective syntheses of these compounds has become the focus of process chemistry initiatives.1–5)
While crystallization from solution is an established technique for the separation and purification of organic compounds, challenges arising from the complex interactions of variables including polymorphism, crystal nucleation and growth, and impurity profile are common.6) For drug substances, understanding and controlling the polymorphism, particle size, morphology, and crystal habit have become fundamental steps in pharmaceutical development programs.7) Crystal polymorphism is one of the most important physicochemical properties in pharmaceuticals. Polymorphic forms have their own characteristics of solubility, bioavailability, and physical/chemical stability. Several techniques for solid-state measurement have been reported in the study of polymorph: powder X-ray diffractometry (PXRD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), microscopy, etc.
Quinolone 006 ((S)-9-fluoro-10-(4-hydroxypiperidin-1-yl)-3-methyl-7-oxo-3,7-dihydro-2H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid L-arginine tetrahydrate) has been under development as a novel quinolone antibiotic. 006 (Chart 1) has superior antibacterial activities against multidrug-resistant bacteria including methicillin-resistant Staphylococcus aureus (MRSA) and penicillin-resistant Streptococcus pneumoniae (PRSP). Because 006 is currently in phase 1 clinical trials, there has been an accelerated demand for its active pharmaceutical ingredient (API). Understanding and controlling the polymorphism and impurity profile of drug substances have become fundamental steps in early research and development programs.8) Less effort has been focused on the manipulation of the temperature and vacuum pressure when drying the APIs, which can affect the polymorphic properties of the API.9) To ensure an uninterrupted supply of 006, we describe the development of a multi-kilogram scale linear synthetic route and crystallization process.
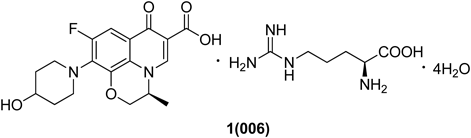
The starting material quinolone core 2 was commercially available. The starting compound 2 was reacted with H3BO4 and Ac2O catalyzed by ZnCl2 to give the chelation intermediate 3 in 94% isolated yield. Compound 3 was reacted with piperidin-4-ol in the presence of Et3N to give the intermediate 4. The chelation group of 4 was deprotected in 5% aqueous acetic acid solution to provide the free acid 5. Then compound 5 was reacted with L-arginine in an acetone/water mixture to give quinolone (S)-9-fluoro-10-(4-hydroxypiperidin-1-yl)-3-methyl-7-oxo-3,7-dihydro-2H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid L-arginine tetrahydrate 1 (006). The scale-up route is shown in Chart 2, while the optimal crystallization and workup process are also described.

Reagents and conditions: (a) H3BO3, Ac2O, ZnCl2, 60°C, 94%; (b) piperidin-4-ol, Et3N, CH3CN, 65°C, quantitative; (c) 5% aqueous AcOH, 80°C, 88%; (d) L-arginine, 53°C; then 5°C crystallization, 86%.
The starting compound 2 was reacted with H3BO4 and Ac2O catalyzed by ZnCl2 to give the chelation intermediate 3.10,11) The initial work-up procedure to provide compound 3 used an ethyl acetate extraction protocol, but the yield was only 66%. Instead, we monitored compound 2 until it was completely consumed (by HPLC ≤ 1%), concentrated the mixture under reduced pressure, then added an ice/water mixture to a stirred solution at 0–5°C to quench the residual Ac2O. The H3BO4 and ZnCl2 were also washed with water during the quenching protocol. The product was isolated in 94% yield and greater than 98% HPLC purity on a 5–10 kg scale.
Synthesis of Coupling Intermediate 4The synthesis of coupling intermediate 4 was carried out by reacting intermediate 3 with piperidin-4-ol in the presence of Et3N in CH3CN at a typical temperature of 65°C. The reaction was worked up by extraction as described previously.12,13) The product was isolated in quantitative yield and greater than 98% purity by HPLC on a 10–15 kg scale.
Synthesis of Free Quinolone Acid 5Because the chelation group is not stable in acidic or basic conditions, the coupling intermediate 4 was deprotected using two methods. Under basic conditions,14) the intermediate 4 was dissolved in 2 M NaOH solution and reacted at 80°C. When the reaction was complete as monitored by HPLC (intermediate 4 ≤ 1%), the mixture was added to a concentrated hydrogen chloride aqueous solution, the pH was adjusted between 6.0 and 7.0, and the desired free quinolone acid was achieved in 78% isolated yield. Under acidic conditions,10) the intermediate 4 was reacted with 5% acetic acid aqueous solution at 80°C, then cooled to room temperature, and filtered to provide the free quinolone acid in 88% isolated yield.
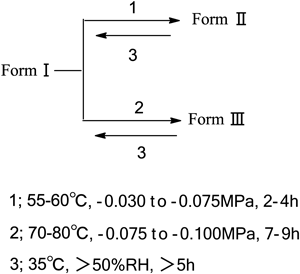
The Synthesis of Target Compound 1 (006)The synthesis of compound 006 was carried out under different conditions. The crystalline polymorphic amino acid salt 1 was prepared by crystallization from the reaction mixture of basic amino acid with compound 5 in an organic–water mixture. We screened different solvents to identify which was the best combination to obtain the L-arginine salt and determined only an acetone–water mixture was suitable for the crystallization. The acetone–water volume ratio required to obtain the desired L-arginine salts was 1 to 1.5. If other organic solvents were added to the acetone–water mixture such as ethanol, i-propyl alcohol or n-propyl alcohol, only (S)-9-fluoro-10-(4-hydroxypiperidin-1-yl)-3-methyl-7-oxo-3,7-dihydro-2H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid L-arginine tetrahydrate (polymorph I) was obtained. Polymorph I, at an ambient temperature of 35°C and relative humidity of 50%, was found to be a tetrahydrate from the elemental analysis. When tetrahydrate 1 was heated at 55 to 60°C for about 2 to 4 h under vacuum (−0.030 to −0.075 MPa), polymorph I was converted to a dihydrate polymorph (polymorph II). When tetrahydrate 1 was heated at 70 to 80°C for about 7 to 9 h under vacuum (−0.075 to −0.1 MPa), polymorph I could lose crystal water and become anhydrate polymorph (polymorph III). However, the dihydrate and anhydrate both reverted to the tetrahydrate form upon exposure to humidity levels higher than 50% (Chart 3). Powder X-ray diffraction (PXRD) (Figs. 1–4) studies of the tetrahydrate, dihydrate and the anhydrate forms confirmed this conclusion. The tetrahydrate polymorph was the most stable hydratomorph of the compound, and was designated 006. The power X-ray diffractometer data of 006 were list in Table 1.

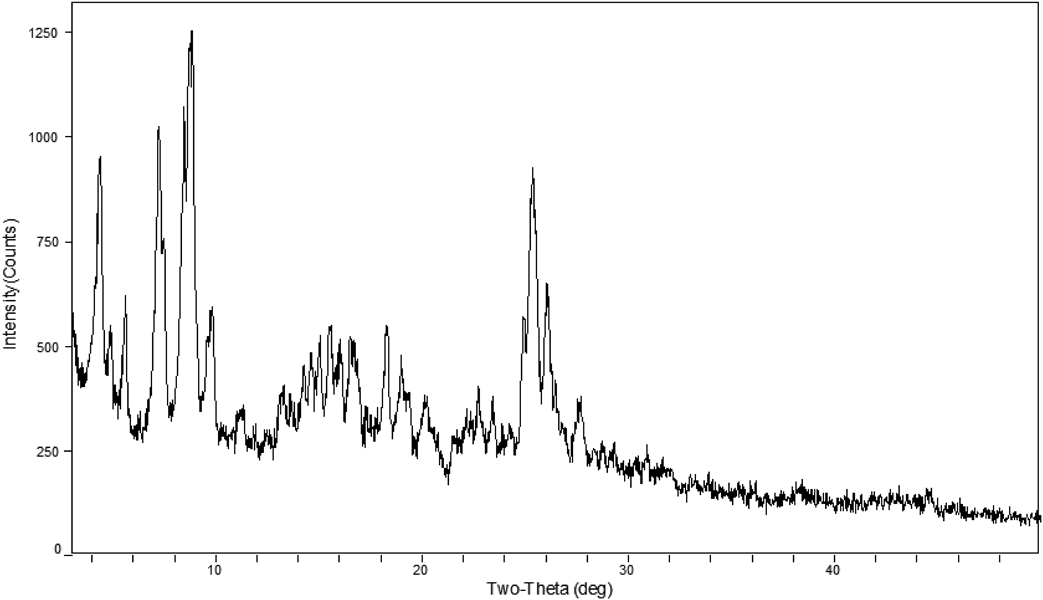


| 2θ° | Interplanar spacing (d) | I/I0 | Crystal density |
|---|---|---|---|
| 4.49 | 19.660 | 28 | 1.168 g/cm3 |
| 4.95 | 17.860 | 30 | |
| 5.68 | 15.571 | 38 | |
| 7.52 | 11.763 | 56 | |
| 8.93 | 9.902 | 100 | |
| 9.66 | 9.149 | 28 | |
| 9.88 | 8.948 | 56 | |
| 16.08 | 5.511 | 19 | |
| 16.54 | 5.361 | 19 | |
| 18.32 | 4.842 | 37 | |
| 19.01 | 4.670 | 25 | |
| 19.39 | 4.579 | 26 | |
| 20.24 | 4.387 | 21 | |
| 24.91 | 3.574 | 35 | |
| 25.44 | 3.501 | 37 | |
| 26.09 | 3.416 | 32 |
After obtaining the tetrahydrate polymorph form of 006, we further optimized the crystallization and workup process. As shown in Table 2, we screened different solvent systems in an effort to optimize the crystallization yield. After dissolving the L-arginine salt in the acetone–water mixture (volume ratio: 1.33), we evaporated the acetone under normal pressure at 60°C, and then slowly added the propan-2-ol (i-PrOH) at 25 and 15°C for a crystallization yield of 39 and 71%, respectively (Table 2, entries 1 and 2). The recrystallization temperature dramatically affected the yield and 15°C was adopted for the recrystallization studies. The acetone–water and ethanol–water systems did not improve the yield at the same temperature (Table 2, entries 3 and 4). Because the two-solvent system did not afford a satisfactory yield, we investigated a three-component system in an effort to improve the yield. Initially, when i-propyl alcohol was added to the reaction mixture, the yield was slightly enhanced to 54% (Table 2, entry 5), but still less than that provided by the i-propyl alcohol–water crystallization system. When we increased the i-propyl alcohol proportion from 72 to 77%, the yield was not noticeably enhanced (Table 2, entry 6). Use of an ethanol–water or ethanol–acetone–water mixture also did not provide sufficient yield. Finally, when the n-propyl alcohol was selected as the anti-solvent, the yield increased to 79% (Table 2, entry 8). To our surprise, when the crystallization temperature cooled to 5°C, the yield got to 86% (Table 2, entry 9), with a purity greater than 99%. It was interesting that when the two-component crystallization solvent systems were employed, a lamellar crystal polymorph habit was observed, while the use of the three-component solvent systems promoted a cluster crystal polymorph habit (Figs. 5, 6).
| Entry | Solvent ratio (v/v) (%) | Temperature (°C) | Time (h) | Yield (%) | Polymorph habits |
|---|---|---|---|---|---|
| 1 | i-PrOH–H2O = 80 : 20 | 25 | 2 | 39 | Lamellar crystal |
| 2 | i-PrOH–H2O = 80 : 20 | 15 | 2 | 71 | Lamellar crystal |
| 3 | Acetone–H2O = 80 : 20 | 15 | 2 | 52 | Lamellar crystal |
| 4 | EtOH–H2O = 80 : 20 | 15 | 2 | 37 | Lamellar crystal |
| 5 | i-PrOH–acetone–H2O = 72 : 16 : 12 | 15 | 2 | 54 | Cluster crystal |
| 6 | i-PrOH–acetone–H2O = 77 : 13 : 10 | 15 | 2 | 56 | Cluster crystal |
| 7 | EtOH–acetone–H2O = 72 : 16 : 12 | 15 | 2 | 45 | Cluster crystal |
| 8 | n-PrOH–acetone–H2O = 72 : 16 : 12 | 15 | 2 | 79 | Cluster crystal |
| 9 | n-PrOH–acetone–H2O = 72 : 16 : 12 | 5 | 2 | 86 | Cluster crystal |
a) All the reactions were carried out at 53°C. The salt formation concentration was 0.26 mol/L. For all experiments, the salt formation solvent ratio (acetone–H2O (v/v)) was 1.33.


The only impurity in the API was the hydrolysis product of the starting material 2 (Fig. 7), which was detected by LC-MS. It was observed as an MH+ ion at a mass of 282.2 (positive ESI source). The exact mass was in accordance with the hydrolysis product of the starting material 2.

In summary, we described the first scale-up synthesis of quinolone 1. Salt formation is a key step in the practical synthesis of the potent antibacterial 006. We also screened the crystallization conditions of the salt formation reaction. The optimal conditions gave 86% isolated yield and greater than 99% purity. Thus, the compound 006 was prepared in 71% yield over 4 steps using a linear route that is convenient, scalable, and does not require a chromatographic purification step.
Unless otherwise indicated, all commercially available reagents were purchased from Aldrich Chemical Company, Inc. (U.S.A.) and used without further purification. Analytical grade solvents were obtained from commercial suppliers (Aldrich and Fisher Scientific, U.S.A.). 1H-NMR, 13C-NMR spectra were recorded on Bruker AVANCE AV 400 (400 MHz) spectrometer. Chemical shifts are reported in ppm related to tetramethylsilane as the internal standard. Element analysis experiments were carried out on an Elementar Vario EL Cube instruments. Powder X-ray diffraction (PXRD) patterns were collected with an Empyrean X-ray powder diffractometer equipped with a real-time-multiple-strip detector, using Ni filtered CuKα radiation at 40 kV, 40 mA. The diffractometer employs a divergence slit of 1/32°, an anti-scatter slit of 1/16°, and soller slits of 0.02 rad at both the incident and the diffracted sides. Each sample was scanned between 3–50°2θ. Thermal properties of the samples were characterized by differential scanning calorimetry (DSC 204, NETZSCH Instruments, Germany). Heating rate of 10°C/min was employed over a temperature range of 25–200°C unless otherwise indicated. Standard aluminum pans were used. The thermogravimetric analysis (TGA) were characterized by thermogravimetric analysis instruments (TG209, NETZSCH Instruments, Germany). Heating rate of 10°C/min was employed over a temperature range of 25–350°C unless otherwise indicated. Standard aluminum pans were used. Melting points were determined with a Yamato MP-21 melting point apparatus in an unsealed capillary tube without correction. Optical rotations were measured in a 1dm cell at 25°C at 589 nm with a WZZ-2B polarimeter. Karl Fisher titrations for water content determination were performed using a Mettler Toledo C20 Coulometric KF titrator. An Aquastar Combi Coulomat Methanol Solution was used as the solvent. Between 10–20 mg of sample was used for each analysis. HPLC measurements were carried out on an HPLC system Agilent-1260 with autoinjector and variable wavelength detector. The chromatography was performed using a 5 µm particle size C18 column (Shiseido Co., Ltd., Japan). Mobile phase used 0.05% trifluoroacetic acid in water and acetonitrile. The detection was performed at 254 nm. HPLC analysis was performed using a Capcell PAK C18 MG S5, 250 × 4.6 mm, 5 µm HPLC column. The mobile phase consisting of a mixture of 0.05% trifluoroacetic acid in water and acetonitrile in the ratio of 68 : 32 was used. The flow rate was at 1 mL/min.
Bis(acetato-O)[(3S)-9,10-difluoro-2,3-dihydro-3-methyl-7-dihydro-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylato-O6,O7]boron (3)A mixture of boric acid (4.8 kg, 77.7 mol), Ac2O (22 L, 232.7 mol), and ZnCl2 (0.088 kg, 0.646 mol) was stirred at room temperature for 30 min. Compound 2 (8.0 kg, 25.9 mol) was added to the reaction, and the mixture was stirred at 60°C for 2 h, and then concentrated in vacuo. The residue was added to an ice–water mixture (160 L) at 0°C, and then stirred at the same temperature for 0.5 h. The product was filtered and washed with water (20 L) and ethanol (20 L), and then the product was dried at 50°C for 5 h to provide 9.9 kg (94%) of intermediate 3. mp: >300°C. [α]D20 +38.66° (c = 0.050, CH2Cl2). 1H-NMR (400 MHz, DMSO-d6) δ: 9.63 (1H, s), 8.01 (1H, dd, J = 10.0, 7.5 Hz), 5.32 (1H, q, J = 6.6 Hz), 4.81 (1H, dd, J = 11.6, 1.3 Hz), 4.65 (1H, dd, J = 11.6, 1.9 Hz), 1.91 (3H, s), 1.91 (3H, s), 1.60 (3H, d, J = 6.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ: 171.3, 168.7, 159.4, 151.7, 151.6, 149.2, 149.1, 147.0, 143.9, 143.8, 141.4, 141.2, 136.6, 136.5, 126.3, 117.3, 117.2, 108.6, 103.1, 102.9, 68.9, 57.2, 22.8, 18.1. Anal. Calcd for C17H14BF2NO8: C, 49.91; H, 3.45; N, 3.42. Found: C, 49.98; H, 3.55; N, 3.53.
Bis(acetato-O)[(3S)-9-fluoro-10-(4-hydroxypiperidin-1-yl)-2,3-dihydro-3-methyl-7-dihydro-7H-pyrido[1,2,3-de][1,4]benzoxazine-6-carboxylato-O6,O7]boron (4)A mixture of intermediate 3 (9.0 kg, 22.0 mol), 4-hydroxypiperidin-1-yl (3.3 kg, 33.0 mol), triethylamine (2.7 kg, 26.4 mol) and CH3CN (132.3 L) was stirred at 65°C for 3.5 h. The mixture was concentrated under reduced pressure. Dichloromethane (135.0 L) and water (135.0 L) were added to the residue, the phases were separated, and the organic phase was washed with saturated NaCl solution (135.0 kg), dried over with Na2SO4 for 20 min, and filtered. The filtrate was concentrated under reduced pressure to give 10.7 kg (quantitative yield) of intermediate 4, which was used directly in the next step without further purification.
(S)-9-Fluoro-10-(4-hydroxypiperidin-1-yl)-3-methyl-7-oxo-3,7-dihydro-2H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic Acid (5)A mixture of intermediate 4 (10.7 kg, 22.0 mol) and 5% acetic acid aqueous solution (v/v, 146.0 L) was stirred at 80°C for 2.5 h. The mixture was cooled to 25°C, the suspension was filtered, and the wet cake was washed with water (18 L) and then ethanol (18 L) to give the wet product. The wet cake was triturated with ethanol (36 L) at 40°C, filtered, and the product was dried at 50°C for 5 h to provide 7.0 kg (88%) of intermediate 5. mp: 194–198°C. [α]D20 −75.60° (c = 0.092, 1 mol/L NaOH). 1H-NMR (400 MHz, DMSO-d6) δ: 15.21 (1H, s), 8.94 (1H, s), 7.56 (1H, d, J = 12.3 Hz), 4.91 (1H, q, J = 6.7 Hz), 4.68 (1H, s), 4.57 (1H, dd, J = 11.5, 1.7 Hz), 4.37 (1H, dd, J = 11.4, 2.2 Hz), 3.73–3.59 (1H, m), 3.52–3.36 (2H, m), 3.23–3.05 (2H, m), 1.84 (2H, dd, J = 8.4, 3.9 Hz), 1.60–1.47 (2H, m), 1.45 (3H, d, J = 6.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ: 176.3, 166.0, 156.7, 154.2, 146.0, 140.0, 132.7, 132.6, 124.7, 119.3, 119.2, 106.5, 103.2, 103.0, 67.9, 65.8, 54.7, 48.3, 35.1, 17.8. Anal. Calcd for C18H19FN2O5: C, 59.66; H, 5.29; N, 7.73. Found: C, 59.60; H, 5.18; N, 7.69.
(S)-9-Fluoro-10-(4-hydroxypiperidin-1-yl)-3-methyl-7-oxo-3,7-dihydro-2H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic Acid L-Arginine Tetrahydrate (1)A mixture of intermediate 5 (6.6 kg, 18.3 mol) and acetone (40 L) was stirred at 25°C for 10 min, and then the L-arginine (3.2 kg, 18.3 mol) and water (30 L) were added to the above solution. The mixture was reacted at 53°C for 1 h until it was a clear solution. The clear solution was added to activated carbon (1 kg) and then stirred at the same temperature for 30 min, cooled to 25°C, and then filtered. The filtrate was added to n-propyl alcohol (180 L) and cooled to 5°C, stirred for 3 h, then filtered. The wet cake was washed with acetone (30 L), then the product was dried at 55–60°C for 4 h under normal pressure to provide 9.6 kg (86%) of compound 1 (006). mp: 159–163°C. [α]D20 −53.72° (c = 0.100, 2% (v/v) ammonia water methanol). 1H-NMR (400 MHz, DMSO-d6) δ: 8.79 (1H, s), 7.95 (4H, s), 7.52 (1H, d, J = 12.5 Hz), 7.16 (2H, s), 4.87–4.77 (1H, m), 4.54 (1H, dd, J = 11.4, 1.7 Hz), 4.34 (1H, dd, J = 11.3, 2.2 Hz), 3.65 (2H, ddd, J = 12.8, 8.6, 3.8 Hz), 3.44–3.35 (3H, m), 3.18–2.95 (6H, m), 1.83 (2H, dd, J = 7.9, 3.7 Hz), 1.63–1.45 (6H, m), 1.43 (3H, d, J = 6.7 Hz). 13C-NMR (100 MHz, DMSO-d6) δ: 176.5, 175.2, 166.9, 157.5, 156.4, 153.9, 145.3, 140.0, 139.9, 131.7, 124.5, 120.5, 103.2, 102.9, 67.9, 65.7, 54.5, 54.0, 48.3, 35.0, 31.0, 25.1, 17.7. Water content: 11.86%. Anal. Calcd for C24H41FN6O11: C, 47.36; H, 6.79; N, 13.81. Found: C, 47.51; H, 6.88; N, 14.00.
We thank the Program of the Pearl River Young Talents of Science and Technology in Guangzhou, China (2013J2200056) for financial support.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.