Abstract
To develop potent ligands for the vitamin D receptor (VDR), we designed and synthesized a series of vitamin D analogues with and without 22-alkyl substituents. These analogues exhibited agonistic, partial agonistic, or antagonistic activity. To elucidate the mechanism of action of the analogues, we conducted crystal structure analyses of the ligand-binding domain (LBD) of VDR complexed with the analogues. The VDR-LBD/agonist complex exhibited precise interactions, which clearly explained VDR agonism. The VDR-LBD/partial agonist complex showed two conformers (agonist and antagonist binding conformers) in a single crystal, demonstrating that partial agonism could be explained by the sum of the agonistic and antagonistic activities. Antagonist binding to the VDR-LBD structure was elucidated using both crystal structure analysis and in-solution structural analyses with the small-angle X-ray scattering (SAXS)-molecular dynamics (MD) and hydrogen/deuterium exchange coupled with mass spectrometry (HDX-MS) methods. Several antagonist-binding structures were detected. We found that the antagonist binding structures differed depending on the structure of the antagonist itself, and those structures clearly explained the VDR antagonism. Furthermore, the apo VDR-LBD structure without the ligand in the ligand-binding pocket was revealed and found to have an entrance to accommodate the ligand. Thus we elucidated the mechanisms of action of agonists, partial agonists, and antagonists based on structural changes (differences) in the receptor protein induced by ligand binding.
Introduction
Calcitriol [1,25-(OH)2D3], an active form of vitamin D, is a hormone that regulates calcium metabolism and plays roles in a variety of physiologic processes, such as cell differentiation induction/proliferation suppression, and immune regulation.1,2) The activity of 1,25-(OH)2D3 is exerted by binding to the vitamin D receptor (VDR) to control the transcription of target genes.3) The VDR is a member of the nuclear receptor family of transcription factors. Nuclear receptors function in gene transcription related to life-critical processes such as development, maintenance of homeostasis, and metabolism.4,5) There are 48 known nuclear receptors in humans, including various steroid hormone receptors, thyroid hormone receptors, retinoid receptors, VDR, and peroxisome proliferator-activated receptors (PPARs). Nuclear receptors become activated by ligand binding, leading to regulation of the transcription of target genes, and this mechanism is common to nuclear receptors.6)
In this study, we designed and synthesized various vitamin D derivatives and studied their interactions with the VDR. We also examined the synthesis of oxidized long-chain fatty acid derivatives and their interactions with the PPAR. We identified a number of agonists, partial agonists, and antagonists and elucidated their mechanisms of action using structural biological methods. Here, we describe our results obtained over the last decade.
1. 22-Alkyl Vitamin D Analogues
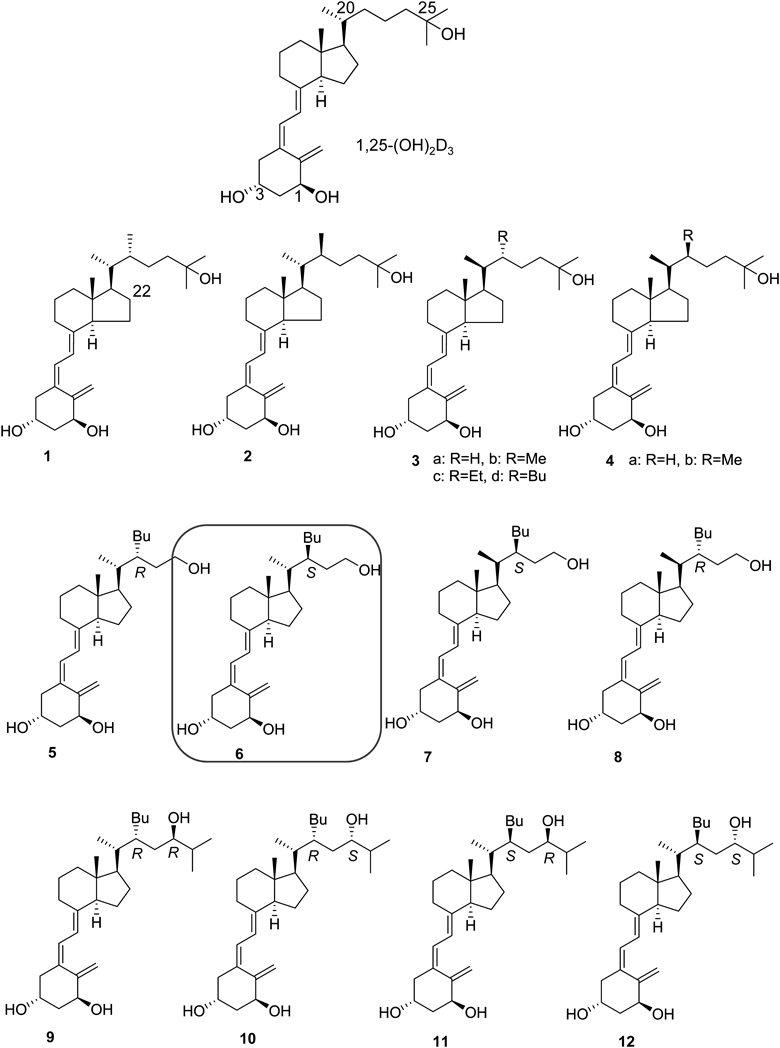
1.1. 22-Butyl-1α,24-dihydroxyvitamin D3 DerivativesMore than 20 years ago, we reported the synthesis of 1,25-(OH)2D3 analogues 1–4 as side-chain-restricted analogues7,8) and found that some 22-alkyl analogues (3b–d) exhibited strong agonistic activity for the VDR7–10) (Fig. 1). Analyses of docking to the ligand-binding domain (LBD) of the VDR and mutational analysis of the VDR suggested that the 22-alkyl substituent of potent analogues 3b–d generates an extra pocket in the ligand-binding pocket (LBP) of the VDR.10,11) Because we were interested in this extra pocket, we considered the synthesis of 22-alkyl analogues. We knew that migration of the hydroxyl group from the 25- to 24-position had minimal effect on agonistic activity for the VDR. Thus we designed and synthesized a series of 22-butyl-1α,24-dihydroxyvitamin D3 derivatives (5–12) to probe the pocket structure of the VDR and evaluated the derivatives’ biological activities12) (Fig. 1). Interestingly, only compound 7 exhibited full agonistic activity, while surprisingly, four compounds, 6, 8, 10, and 12, inhibited transactivation induced by 1,25-(OH)2D3, indicating that these compounds are VDR antagonists but not agonists. Other compounds, 5, 9, and 11, did not exhibit significant activity.
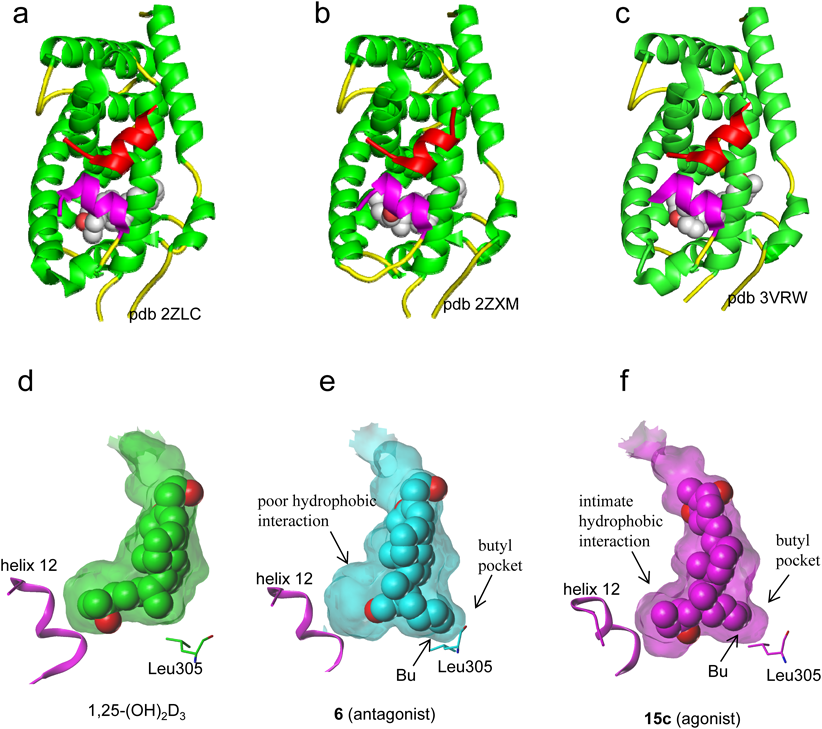
All crystal structures of the VDR-LBD/ligand complex that had been reported up to that time adopted the active conformation13,14) (Figs. 2a, d), and there were no crystal structures to explain VDR antagonism. To elucidate the mechanism of antagonism of the VDR, we attempted to crystallize the VDR-LBD/antagonist complex and succeeded in crystallization of the VDR-LBD/antagonist 6 complex. The overall structure of the VDR-LBD/6 complex adopted the active conformation12) (Fig. 2b). We considered that the structure did this by effects on crystal packing. However, an extra pocket to accommodate the 22-butyl group of compound 6 was discovered12) (Fig. 2e), which we designated a “butyl pocket.” In addition, the complex revealed that ligand 6 forms poor hydrophobic interactions with helix 12 (Fig. 2e). From these results, we hypothesized that both butyl pocket formation and the weak hydrophobic interactions with helix 12 are related to the antagonistic behavior to the VDR.7–12,15) From a series of studies, we identified VDR ligands with a simple side chain that forms the butyl pocket and hypothesized that the 22-alkyl substituent of the analogue would modulate the butyl pocket structure. We concluded that crystal structure analysis of the VDR-LBD/22-alkyl analogue complex would be useful for enhancing our understanding of the mechanism of VDR antagonism.
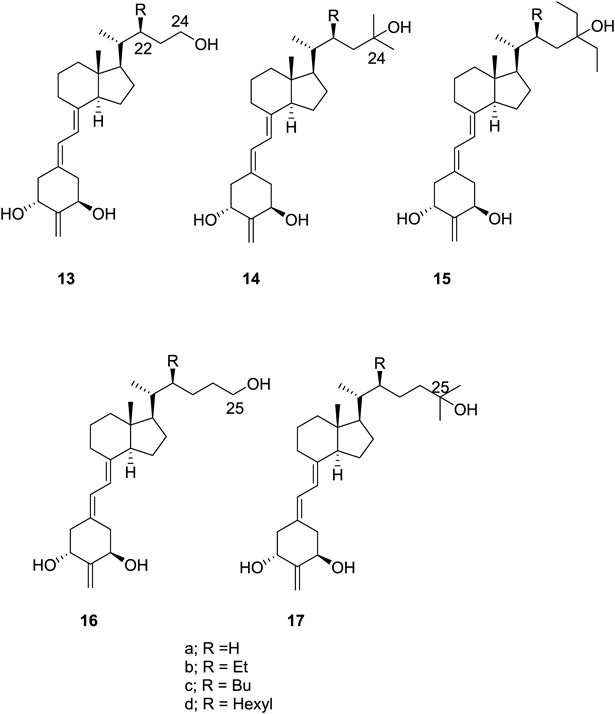
1.2. Vitamin D Analogues with and without a 22S-Alkyl SubstituentAs described above, we found that the 22S-butyl substituent of compound 6 induces formation of the butyl pocket in the VDR and that ligand 6 interacts poorly with helix 12 of the VDR-LBD. Therefore to investigate the effects of butyl pocket formation and poor interaction with helix 12 on VDR agonism/antagonism, a series of vitamin D3 analogues with and without a 22S-alkyl substituent (13a–d, 14a–c, 15a–c, 16a–c, 17a–c) were designed and synthesized and their biological activity examined16,17) (Fig. 3). Interestingly, the activity of each synthetic compound varied depending on the side-chain structure. Thus 13a, 14a, 15a–c, 16a, and 17a–c exhibited agonistic activity, whereas 13b, d 14b, c, and 16b exhibited partial agonistic activity, and 13c, 16c exhibited antagonistic activity.
Unfortunately, analyses of the crystal structure of the VDR-LBD complexed with antagonists were unsuccessful; however, analysis of the VDR-LBD complexed with the potent agonist 15c was successful17) (Fig. 2c). In the VDR-LBD/agonist 15c complex, the butyl pocket was generated, and helix 12 interacted closely and hydrophobically with ligand 15c (Fig. 2f). These results indicate that both the generation of the butyl pocket in the VDR and poor hydrophobic interaction with VDR helix 12 are crucial for function as a VDR antagonist. These two structural factors must contribute to inhibit the active conformation of helix 12.
From the above studies, we confirmed the following. Among ligands inducing formation of the butyl pocket (6, 13c, and 15c), the ligands that do not interact with the VDR C-terminus (6, 13c) function as antagonists, whereas the ligand that does interact closely with the VDR C-terminus (15c) functions as an agonist. In addition, ligands that do not induce butyl pocket formation (13a, 14a, and 15a) enhance agonist activity if the ligand enhances interactions with the VDR C-terminus. Thus the activity of the ligand as a VDR agonist or antagonist is strongly affected by formation of the butyl pocket in the VDR. It should be noted from the above studies that the VDR structure to explain antagonism remains unclear yet.
2. Mechanism of Partial Agonist
2.1. Vitamin D Analogues with 22R-Alkyl SubstituentsVitamin D analogues with alkyl substituent of the S-configuration at the 22-position were found to act as an agonist, partial agonist, or antagonist for the VDR. These analogues were also found useful for probing the pocket structure of the VDR, as described above. Next, we were interested in determining the potency of counterpart analogues with an alkyl substituent of the R-configuration at the 22-position. Therefore we designed and synthesized a series of 22R-alkyl analogues (18a, b–22a, b) and examined their biological activity18) (Fig. 4). All compounds bound to the VDR, but only 22a exhibited agonistic activity. Compounds 18a, 21a, b, and 22b exhibited partial agonistic activity, and the remaining compounds, 18b, 19a, b and 20a, b, exhibited antagonistic activity. Crystallographic analyses of only the VDR-LBD/21a (Fig. 5a) and VDR/22a (data not shown) complexes were successful.18) Analysis of the VDR-LBD/22a complex indicated that the 22R-butyl substituent induced formation of the butyl pocket, similar to the VDR-LBD/15c complex, despite the different configuration of the butyl group at the 22-position (data not shown).
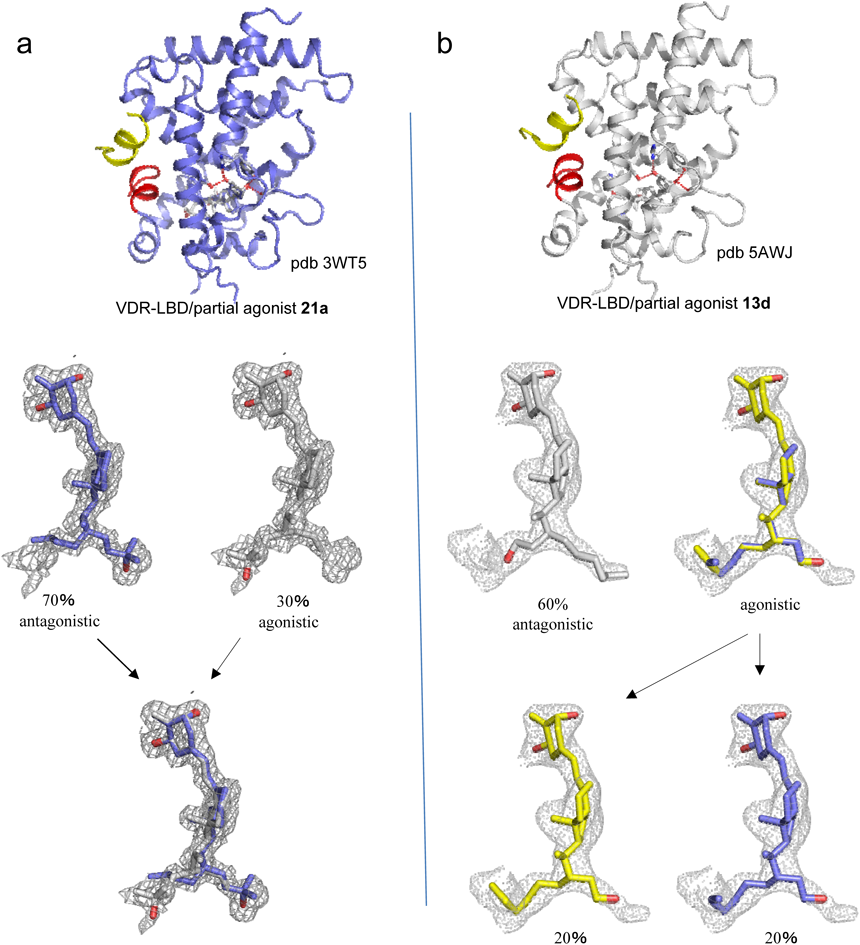
Interestingly, the structure of the VDR-LBD/21a complex yielded valuable data regarding partial agonism of the VDR. Thus a mixed population of antagonist- and agonist-binding conformers was observed in a single crystal at ratios of 70 and 30%18) (Fig. 5a). This observation suggests that the partial agonistic behavior of 22R-butyl analogue 21a is based on the sum of antagonistic and agonistic activities of the antagonist and agonist binding conformers. Taken together with our previous results, the following conclusions concerning agonism, partial agonism, and antagonism of the VDR were derived. When close interactions of the ligand with helix 11/12 of the VDR through hydrogen bonding with His393 and hydrophobic bonding overcomes the structural strain in the VDR caused by formation of the butyl pocket, the ligand functions as an agonist for the VDR. Conversely, when the ligand interacts poorly with helix 11/12 of the VDR, the ligand functions as a VDR antagonist. More interestingly, the sum of agonistic and antagonistic behaviors of the VDR/ligand complex results in VDR partial agonist activity. The mechanism of partial agonism derived from this study could be suitable for the partial agonism of other nuclear receptors, for example steroid hormone receptors and retinoid hormone receptors.
2.2. Vitamin D Analogue with a 22S-Hexyl Group22S-Hexyl analogue 13d was designed and synthesized under the expectation that it would be a potent VDR antagonist; however, it was found to function as a partial agonist.19) Interestingly, in the crystal structure of the VDR-LBD/13d complex, partial agonist 13d was accommodated in the VDR in two conformations19) (Fig. 5b). The major conformer (60%) was related to antagonistic activity, whereas two minor conformers (each 20%) were related to agonistic activity. Thus the partial agonist activity of compound 13d could be explained by the mixed population of antagonistic and agonistic conformers.19) This is the second instance of detecting both agonistic and antagonistic conformations in a single crystal and explaining the partial agonism by the contribution of a mixed population of both conformations.
Although it was found that the antagonistic activity increased in the order 13a (H) < 13b (Et) < 13c (Bu), compound 13d (Hex) exhibited agonistic activity.19) This study demonstrates that the length of 22-alkyl group of the ligands allows for fine tuning of agonistic/antagonistic activity against the VDR. This finding is extremely useful for the development of novel VDR agonists/antagonists.
3. Apo- and Antagonist-Binding Structures of the VDR in Solution
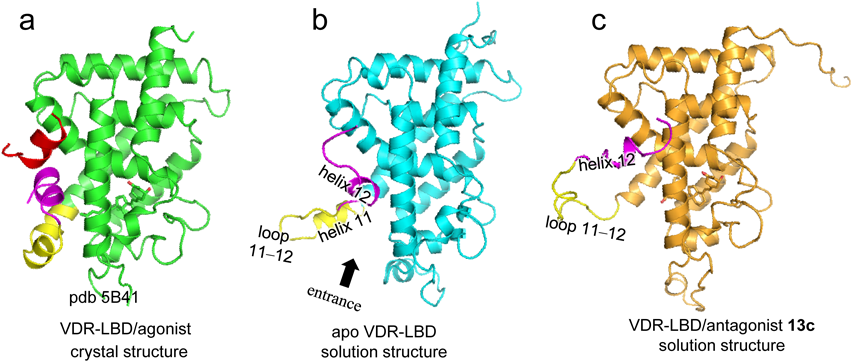
The crystal structure of apo VDR-LBD, in which the specific ligand is absent, has not been reported, and the crystal structure of the VDR-LBD/antagonist complex, which explains VDR antagonism, remains unclear. It is thought that the reason these structures have not been fully solved is related to fluctuation of the apo VDR-LBD and the VDR-LBD/antagonist complex being too large for adequate crystallization. We therefore plan to analyze the in-solution structures—rather than the crystal structures— of apo VDR-LBD and the VDR-LBD/antagonist complex.
3.1. Protein Structure in Solution (SAXS-MD)Small-angle X-ray scattering (SAXS) analysis can be used to elucidate the overall shape of the structure of a macromolecule under almost any physiologic aqueous condition.20–23) We attempted to analyze the in-solution structures of apo VDR-LBD and the VDR-LBD/antagonist complex using SAXS, but the structures could not be clarified because they were too different from known VDR-LBD structures. Therefore further SAXS analyses were performed with the aid of molecular dynamics (MD) simulations, thus combining SAXS and MD.
3.2. Apo- and Antagonist-Binding StructuresThe apo- and antagonist-binding structures of the VDR-LBD were revealed using a SAXS-MD hybrid approach.24) In-solution structures of apo VDR-LBD (Fig. 6b) and the VDR-LBD/antagonist complex (Fig. 6c) are shown compared with the crystal structure of the VDR-LBD/agonist complex (Fig. 6a). The structure of apo VDR-LBD was almost identical to the agonist-binding structure of the VDR-LBD up to helix 10, whereas the structure from the end of helix 11 to the beginning of helix 12 was disordered (Fig. 6b). Helix 11 turns outward at the kink position, resulting in opening of the entrance for the ligand into the LBP. Helix 12 is partially unraveled, fluctuates, and rotates toward the coactivator binding site. This structure clearly describes the characteristics of apo structure, which although normally unstable, is stabilized by ligand binding.
In VDR-LBD/antagonist complex 13c, the structure was nearly identical to the agonist-binding structure up to helix 10, whereas the structure of helix 11 and the subsequent C-terminus was largely perturbed (Fig. 6c). Thus partial unraveling of helixes 11 and 12 revealed the wide and flexible loop 11–12 between helixes 11 and 12. Helix 12 is positioned near the canonical coactivator binding region, so that formation of the active conformation surface is inhibited, thus preventing coactivator binding. This structure clearly explains the mechanism of VDR antagonism.
Because the side chain structure of antagonist 13c is the same as that of antagonist 6, the mechanism of antagonistic action of both compounds is considered the same. Therefore in-solution structure of VDR-LBD/6 complex is thought to adopt the same structure as the VDR-LBD/13c complex obtained in this study. Thus by using SAXS-MD hybrid approach, we first revealed the structure of VDR-LBD/antagonist complex, which explains the VDR antagonism.
4. Antagonist-Binding Structures of VDR
4.1. Protein Dynamics in Solution (HDX-MS)Hydrogen/deuterium exchange coupled with mass spectrometry (HDX-MS) has been used to study protein–protein interactions, protein folding, and protein–ligand interactions.25–28) The static state is elucidated using X-ray crystal structure analysis, whereas HDX-MS analysis elucidates protein dynamics in solution. HDX-MS analysis of proteins shows the rate of hydrogen/deuterium exchange of backbone amide hydrogens, disclosing the strength of hydrogen bonds, conformational mobility, and the accessibility of solvent.25,26)
4.2. Structure That Destabilizes Helix 6–7Vitamin D analogues 23a, b–27a, b were designed and synthesized, and their biological activity was evaluated, so as to develop strong VDR antagonists based on the mechanism of inhibition of helix 12 folding caused by the bulky side chain of the ligand. This study demonstrated that the 25-dibenzyl analogues 24a and 24b both function as potent VDR antagonists29) (Fig. 7). HDX-MS analysis showed that 1,25-(OH)2D3 strongly stabilizes the region forming the LBP (Fig. 8a). Antagonist 24a had significant stabilizing effect on the LBP region (Fig. 8b); however, unexpectedly, 24a strongly destabilized helix 6–7 but strongly stabilized helix 11–12 (Fig. 8b). Analysis of the X-ray crystal structure also showed that helix 6–7 of the VDR-LBD complexed with 24a is shifted from the canonical position, whereas helix 11–12 adopts the same conformation as the agonist-binding conformation (Figs. 8d, e). At that time, no antagonist that strongly stabilizes helix 12 was known; therefore this was the first study to identify such an antagonist. This type of antagonist should be identified for other nuclear receptors as well.
In contrast, HDX-MS analysis indicated that compound 24b destabilizes helix 11–12 (Fig. 8c); therefore 24b is considered an antagonist that inhibits folding of helix 12. In this study, we demonstrated that VDR antagonists function via different mechanisms dependent on their structure. This study also demonstrated the usefulness of HDX-MS for probing changes in protein structures caused by ligand binding.
4.3. Structures That Destabilize Helix 11–12The abovementioned study showed that VDR-LBD complexed with antagonist 24a adopts an alternative conformation at helix 6–7. This interesting finding was confirmed using HDX-MS and crystal structure analyses. Subsequently, a similar conformational alteration of helix 6–7 was reported by Shimizu and colleagues30) using crystal structure analysis. However, these crystal structure analyses did not reveal any alternative conformation of helix 11–12.
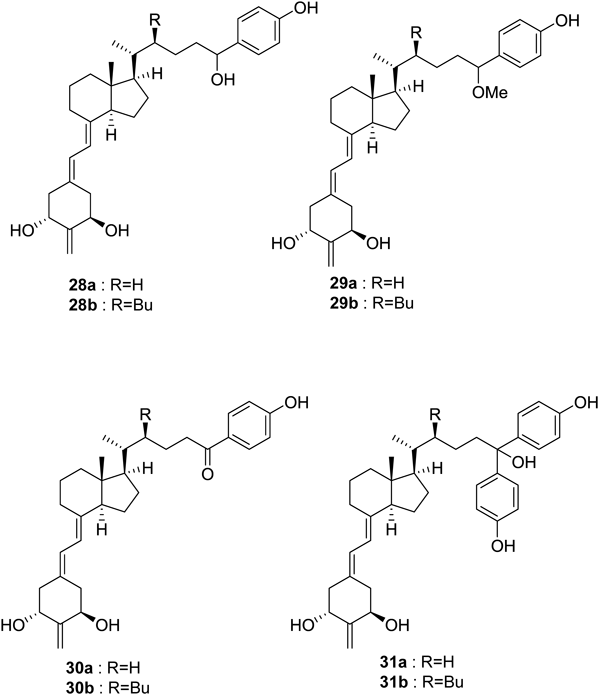
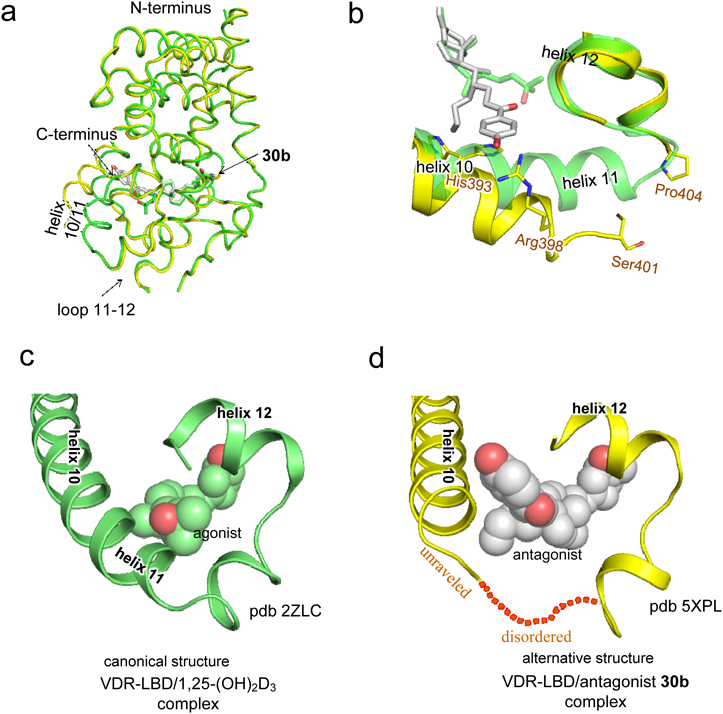
To obtain a crystal structure of an alternative conformation of helix 11–12, we continued to develop appropriate antagonists. We designed and synthesized vitamin D analogues with a p-hydroxyphenyl group at the side-chain terminus (28a, b–31a, b) and evaluated their biological activities31) (Fig. 9). All these compounds bound to the VDR, but only 29a exhibited agonistic activity. Compounds 28a, b, 29b, and 30a exhibited partial agonistic activity, and the remaining compounds, 30b and 31a, b, exhibited antagonistic activity. We attempted to crystalize the VDR-LBD complexed with antagonist 30b, 31a, or 31b and succeeded in the crystallographic analysis of the VDR-LBD/30b complex. The overall structure was almost identical to the VDR-LBD/1,25-(OH)2D3 complex up to the beginning of helix 10, but significant differences at helix 10/11 were apparent31) (Fig. 10). In the VDR-LBD/30b complex, no electron density was observed along the unraveled part of helix 11 (Fig. 10a, b, d). Furthermore, a significant shift of the main chains of helix 10/11 relative to the agonist binding structure14) was observed (Figs. 10a, b). This structural change was considered caused by steric repulsion of the p-hydroxyphenyl group of 30b with a His residue on helix 10.31) Thus we presented the first report clearly demonstrating a different crystal structure conformation of the C-terminus region helix 10–12 relative to the agonist binding structure (Figs. 10c, d). The present study indicates that antagonists for nuclear receptors, including VDR, modulate the protein structure at various positions, suggesting the possibility of designing novel antagonists that modulate positions of the protein distinct from previously known positions.
5. PPARγ Ligand with Partial Agonistic Activity
We chemically synthesized a series of oxidation products of highly unsaturated long-chain fatty acids such as docosahexaenoic acid and eicosapentaenoic acid and found many of them exhibited agonistic or partial agonistic activity for PPARγ.32–41) We therefore conducted a crystal structural analysis of PPARγ-LBD accommodating an oxidized fatty acid. Figure 11 summarizes analysis of the crystal structure of the PPARγ-LBD complexed with 5-oxo-tricosahexaenoic acid (5-oxoTrHA) and 6-oxo-tetracosahexaenoic acid (6-oxoTHA), which are partial agonists for PPARγ38) (Fig. 11). In both the PPARγ-LBD/5-oxoTrHA and PPARγ-LBD/6-oxoTHA complexes, PPARγ was packed in the crystals as a homodimer in which monomers A and B adopted active and inactive conformations, respectively38) (Figs. 11a, b). The behavior of this crystal structure was quite similar to that of the VDR-LBD/partial agonist complex.18,19) Therefore the partial agonist activity may be explained by the sum of the agonistic and antagonistic activities based on the agonist and antagonist binding conformers, respectively. Thus in the nuclear receptors VDR and PPARγ, partial agonism is explained by contribution of the agonist and antagonist binding structures, strongly suggesting that partial agonist activity is the result of equilibrium between the agonist binding conformer and antagonist binding conformer.
6. Conclusion
Prior to the present study, the mechanism of agonism/antagonism of VDR initiated by ligand binding was poorly understood due to the lack of precise structural elucidation of the apo state and the almost identical crystal structures of all VDR-LBD/ligand complexes, regardless of agonist/antagonist binding. We developed ligands of the VDR and PPAR and elucidated the mechanism of the agonistic, partial agonistic, and antagonistic activities using our synthetic ligands. Figure 12 summarizes the mechanism of VDR agonism, partial agonism, and antagonism as revealed by our research described in this review. All VDR-LBD structures except agonist binding structure shown in Fig. 12 were first detected in this study by X-ray crystallographic analysis and SAXS-MD hybrid analysis. In apo VDR-LBD, the C-terminus from helix 11 to helix 12 fluctuates significantly to provide the ligand entrance to the LBP. When the agonist enters the pocket, the entrance closes to form the stable agonist binding structure of the VDR, ultimately leading to transcription of target genes. When an antagonist enters the pocket, the VDR assumes an antagonist binding structure that inhibits transcription of target genes. We emphasize that there is only one agonist-binding structure, and a variety of antagonist-binding structures that depend on the ligand structure. Partial agonist activity is explained by equilibrium between the agonist binding and antagonist binding structures. We rationally elucidated the mechanisms of action of agonists, partial agonists, antagonists based on changes in the structure (differences) of the receptor protein induced by ligand binding. This mechanism can be applied to that of other nuclear receptors and their ligands.
Acknowledgments
The achievements reported in this review were due to the consistently enthusiastic efforts of talented and devoted former and present co-workers, whose names are cited in the references. These studies were financially supported by the followings: Grants-in-Aid for Scientific Research (Grants 20590108, 23590135, and 26460155) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); a Grant-in-Aid for High Technology Research Center Project (Grant 19-8) from the MEXT; the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2013–2017); the Platform for Drug Discovery, Informatics, and Structural Life Science from the Japan Agency for Medical Research and Development (AMED); and the Takeda Science Foundation, Japan.
Conflict of Interest
The author declares no conflict of interest.
Note
This review of the author’s work was written by the author upon receiving the 2018 Pharmaceutical Society of Japan Award for Divisional Scientific Contribution.
References
- 1) “Vitamin D,” ed. by Feldman D., Pike J. W., Glorieux F. H., 2nd ed., Elsevier Academic Press, New York, 2005.
- 2) Pike J. W., Meyer M. B., Bishop K. A., Rev. Endocr. Metab. Disord., 13, 45–55 (2012).
- 3) Jurutka P. W., Whitfield G. K., Hsieh J. C., Thompson P. D., Haussler C. A., Haussler M. R., Rev. Endocr. Metab. Disord., 2, 203–216 (2001).
- 4) Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R. M., Cell, 83, 835–839 (1995).
- 5) Olefsky J. M., J. Biol. Chem., 276, 36863–36864 (2001).
- 6) Kojetin D. J., Burris T. P., Mol. Pharmacol., 83, 1–8 (2013).
- 7) Yamamoto K., Takahashi J., Hamano K., Yamada S., Yamaguchi K., DeLuca H. F., J. Org. Chem., 58, 2530–2537 (1993).
- 8) Yamamoto K., Sun W.-Y., Ohta M., Hamada K., DeLuca H. F., Yamada S., J. Med. Chem., 39, 2727–2737 (1996).
- 9) Yamamoto K., Ooizumi H., Umesono K., Verstuyf A., Bouillon R., DeLuca H. F., Shinki T., Suda T., Yamada S., Bioorg. Med. Chem. Lett., 9, 1041–1046 (1999).
- 10) Yamamoto K., Inaba Y., Yoshimoto N., Choi M., DeLuca H. F., Yamada S., J. Med. Chem., 50, 932–939 (2007).
- 11) Choi M., Yamamoto K., Itoh T., Makishima M., Mangelsdorf D. J., Moras D., DeLuca H. F., Yamada S., Chem. Biol., 10, 261–270 (2003).
- 12) Inaba Y., Yoshimoto N., Sakamaki Y., Nakabayashi M., Ikura T., Tamamura H., Ito N., Shimizu M., Yamamoto K., J. Med. Chem., 52, 1438–1449 (2009).
- 13) Rochel N., Wurtz J. M., Mitschler A., Klaholz B., Moras D., Mol. Cell, 5, 173–179 (2000).
- 14) Shimizu M., Miyamoto Y., Takaku H., Matsuo M., Nakabayashi M., Masuno H., Udagawa N., DeLuca H. F., Ikura T., Ito N., Bioorg. Med. Chem., 16, 6949–6964 (2008).
- 15) Inaba Y., Nakabayashi M., Itoh T., Yoshimoto N., Ikura T., Ito N., Shimizu M., Yamamoto K., J. Steroid Biochem. Mol. Biol., 121, 146–150 (2010).
- 16) Sakamaki Y., Inaba Y., Yoshimoto N., Yamamoto K., J. Med. Chem., 53, 5813–5826 (2010).
- 17) Yoshimoto N., Sakamaki Y., Haeta M., Kato A., Inaba Y., Itoh T., Nakabayashi M., Ito N., Yamamoto K., J. Med. Chem., 55, 4373–4381 (2012).
- 18) Anami Y., Itoh T., Egawa D., Yoshimoto N., Yamamoto K., J. Med. Chem., 57, 4351–4367 (2014).
- 19) Anami Y., Sakamaki Y., Itoh T., Inaba Y., Nakabayashi M., Ikura T., Ito N., Yamamoto K., Bioorg. Med. Chem., 23, 7274–7281 (2015).
- 20) Amemiya Y., Wakabayashi K., Hamanaka T., Wakabayashi T., Hashizume H., Nucl. Instrum. Methods Phys. Res., 208, 471–477 (1983).
- 21) Ueki T., Hiragi Y., Kataoka M., Inoko Y., Amemiya Y., Izumi Y., Tagawa H., Muroga Y., Biophys. Chem., 23, 115–124 (1985).
- 22) Vestergaard B., Arch. Biochem. Biophys., 602, 69–79 (2016).
- 23) Ogawa T., Hirokawa N., Biophys Rev., 10, 299–306 (2018).
- 24) Anami Y., Shimizu N., Ekimoto T., Egawa D., Itoh T., Ikeguchi M., Yamamoto K., J. Med. Chem., 59, 7888–7900 (2016).
- 25) Wales T. E., Engen J. R., Mass Spectrom. Rev., 25, 158–170 (2006).
- 26) Skinner J. J., Lim W. K., Bédard S., Black B. E., Englander S. W., Protein Sci., 21, 996–1005 (2012).
- 27) Zhang J., Chalmers M. J., Stayrook K. R., Burris L. L., Garcia-Ordonez R. D., Pascal B. D., Burris T. P., Dodge J. A., Griffin P. R., Structure, 18, 1332–1341 (2010).
- 28) Zhang J., Chalmers M. J., Stayrook K. R., Burris L. L., Wang Y., Busby S. A., Pascal B. D., Garcia-Ordonez R. D., Bruning J. B., Istrate M. A., Kojetin D. J., Dodge J. A., Burris T. P., Griffin P. R., Nat. Struct. Mol. Biol., 18, 556–563 (2011).
- 29) Kato A., Itoh T., Anami Y., Egawa D., Yamamoto K., Bioconjug. Chem., 27, 1750–1761 (2016).
- 30) Asano L., Waku T., Abe R., Kuwabara N., Ito I., Yanagisawa J., Nagasawa K., Shimizu T., FEBS Lett., 590, 3270–3279 (2016).
- 31) Kato A., Yamao M., Hashihara Y., Ishida H., Itoh T., Yamamoto K., J. Med. Chem., 60, 8394–8406 (2017).
- 32) Yamamoto K., Itoh T., Abe D., Shimizu M., Kanda T., Koyama T., Nishikawa M., Tamai T., Ooizumi H., Yamada S., Bioorg. Med. Chem. Lett., 15, 517–522 (2005).
- 33) Itoh T., Murota I., Yoshikai K., Yamada S., Yamamoto K., Bioorg. Med. Chem., 14, 98–108 (2006).
- 34) Itoh T., Yamamoto K., Naunyn Schmiedebergs Arch. Pharmacol., 377, 541–547 (2008).
- 35) Yamamoto K., Ninomiya Y., Iseki M., Nakachi Y., Kanesaki-Yatsuka Y., Yamanoue Y., Itoh T., Nishii Y., Petrovsky N., Okazaki Y., Biochem. Biophys. Res. Commun., 367, 566–572 (2008).
- 36) Itoh T., Yoshimoto N., Yamamoto K., Heterocycles, 80, 689–695 (2010).
- 37) Itoh T., Tomiyasu A., Yamamoto K., Lipids, 46, 455–461 (2011).
- 38) Egawa D., Itoh T., Yamamoto K., Bioconjug. Chem., 26, 690–698 (2015).
- 39) Yamamoto Y., Itoh T., Yamamoto K., J. Nutr. Sci. Vitaminol., 61, 222–227 (2015).
- 40) Itoh T., Saito T., Yamamoto Y., Ishida H., Yamamoto K., Bioorg. Med. Chem. Lett., 26, 343–345 (2016).
- 41) Egawa D., Itoh T., Akiyama Y., Saito T., Yamamoto K., ACS Chem. Biol., 11, 2447–2455 (2016).