Reviews
Chemical Studies on Bioactive Natural Products Directed toward Development of Novel Antiinfective and Anticancer Medicines
2019 年 67 巻 7 号 p. 620-631
詳細
2019 年 67 巻 7 号 p. 620-631
Natural products are still rich sources of clinically used medicines and lead compounds for them. This review summarizes chemical studies carried out by the author on natural products of microorganism origin, many of which were discovered at the Institute of Microbial Chemistry (BIKAKEN). Caprazamycin B is a liponucleoside antibiotic from which CPZEN-45, an antituberculosis agent with a unique mode of action, was developed. Intervenolin and leucinostatin A exert antiproliferative activity toward tumor cells in the presence of the corresponding stromal cells, which implies that the primary molecular targets of these molecules should be related to growth signals from normal (stromal) cells. Details of the endeavors to establish efficient synthetic routes to these compounds which accelerated structure–activity relationship studies and further evaluation of biological activity are described.
Nature produces countless biologically active substances that are widely recognized as a rich source of clinically used drugs and their precursors.1–3) In particular, the contribution of microorganisms to the production of antibiotics cannot be overemphasized. It is reasonable that varying species of microorganisms are fighting to survive by maintaining or expanding their territory with antibiotics as warheads; humans benefit by borrowing the weapons in the form of antiinfective agents. Antibiotics are now prescribed to treat diverse diseases and not limited to infections. The structural diversity of natural products expands their opportunities to interact with a variety of biomolecules, which means that previously unknown molecular targets for therapies are expected to be clarified in phenotypic assay systems employing natural products as screening sources. This review article summarizes the author’s efforts to maximize the potential of biologically active natural substances, especially those originating from microorganisms, as medicinal leads based on chemical approaches.
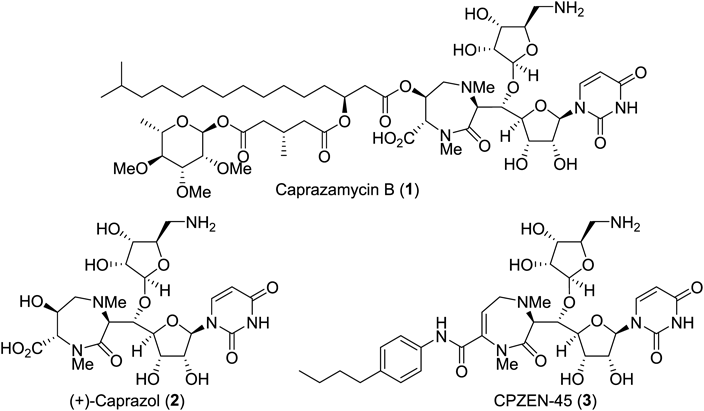
Tuberculosis is still a major threat to public health worldwide, which is currently worsening with the emergence of extensively drug-resistant strains of pathogenic Mycobacterium tuberculosis (XDR-TB). The current lack of efficacious clinical agents against these strains urgently demands drug candidates with novel modes of action. The liponucleoside antibiotic caprazamycin B (1, Fig. 1), characterized by a centrally located seven-membered lactam (diazepanone ring system) connected with a uridine moiety and an aliphatic ester side chain, was first reported by Igarashi et al. in 2003.4,5) Fermentation gave this natural product as a mixture with structurally related substances bearing aliphatic side chains with varying lengths and branching patterns. Each form of caprazamycin displays favorable anti-TB activity, among which caprazamycin B was found to be the most potent. A core structure of caprazamycins, caprazol (2, Fig. 1), which itself is a natural product, showed no antibacterial activity, while CPZEN-45 (3)6) semisynthetically derived from caprazamycins exerts anti-XDR-TB activity. Caprazamycins and liposidomycins, reported by Isono et al. in 1985,7) share characteristic structural features; both molecules exhibit anti-TB activity by inhibiting MraY, one of the key enzymes in the biosynthesis of the peptidoglycan of TB. On the other hand, CPZEN-45 has a distinctive molecular target, WecA,8) an enzyme for the biosynthesis of mycolyl arabinogalactan at the tuberculosis cell wall, which should be the origin of its intriguing anti-XDR-TB property. The unique biological activity and structural novelty of the compounds of this class drew tremendous attention from the synthetic community. The first total syntheses of caprazol (2) were achieved by Matsuda, Ichikawa,9–11) and colleagues, which was followed by the first disclosure of caprazamycin A by Takemoto’s group.12)
Also attracted by these molecules, the author decided to establish synthetic routes to be applied to structure–activity relationship (SAR) studies to develop anti-XDR-TB agents with greater potency and favorable pharmacological characteristics. Considering the structural complexity similar to that of CPZEN-45, caprazol (2) was set as a primary synthetic target.
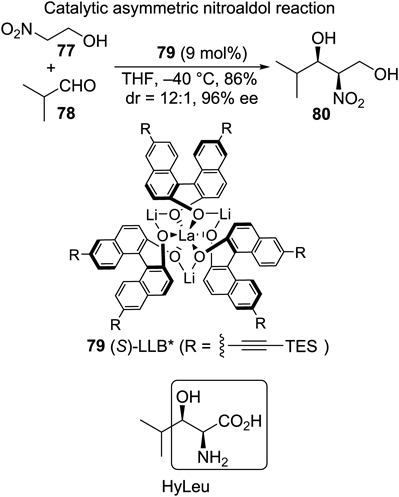
Chart 1 shows the two key stereoselective C–C bond-forming reactions in the total synthesis of caprazol,13) the diastereoselective isocyanoacetate aldol reaction14) employing methyl isocyanoacetate (4) and a uridine-derived aldehyde (5), and the antiselective catalytic asymmetric nitroaldol reaction reported by Nitabaru et al.15–17) using cinnamaldehyde (7) and benzylated nitroethanol (8). In the former, pronucleophile 4 is sufficiently active to undergo deprotonation at the α-position of the carbonyl functionality even with a tertiary amine to afford trans-oxazoline products 6 and 6′ almost exclusively, albeit with low facial selectivity, at 1.2 : 1. Copper-catalyzed conditions reported by Kirchner’s group18) (Chart 1) raise the ratio to as high as 7.3 : 1. In this reaction, structurally hindered protecting groups for the sugar moiety of 5 were essential: e.g., the isopropylidene congener diminished diastereoselectivity to 1 : 1.5 (the undesired isomer predominated). For the second key transformation, the nitroaldol reaction, an amide ligand 9 complexed with Nd and Na metals acts as a bimetallic bifunctional catalyst to activate both the electrophile 7 and pronucleophile 8 simultaneously. During the reaction course, an extended transition state is proposed to be operative to afford the anti-product predominantly. The use of 9 mol% of the catalyst system produced the desired enantiomer 10 in dr of 12 : 1 and 95% enantiomeric excess (ee).
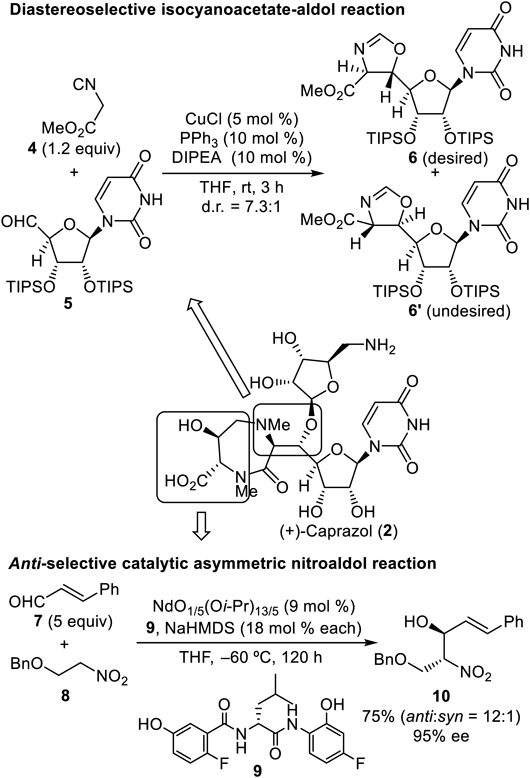
The resultant oxazolidinone ring of 6 was cleaved to give 11 in an acidic medium without damaging the stereochemical preference, which was followed by protection of the primary amine leading to 12 (Chart 2). According to the procedure reported for the total synthesis of caprazol,9–11) the subsequent Lewis acid-promoted glycosylation employing a fluorinated glycosyl donor 13 proceeded successfully.
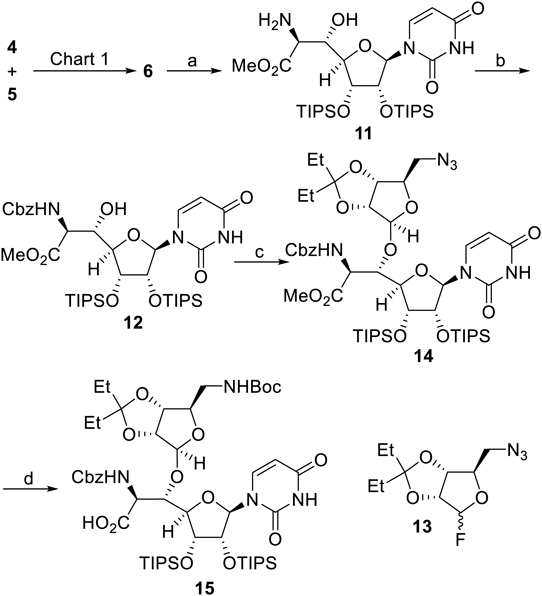
Reagents and conditions: (a) 4 M HCl, THF, room temperature (r.t.), 67%; (b) CbzCl, NaHCO3, THF–H2O (2 : 1), 0°C, 90%; (c) 13, BF3·OEt2, MS 4A, CH2Cl2, −30°C, 72%; (d) i) PPh3, H2O, toluene–THF (1 : 1), 50°C; ii) Boc2O, NaHCO3, r.t., 90% (2 steps); iii) LiI, AcOEt, 80°C, 64%.
In parallel, the product from the catalytic asymmetric nitroaldol reaction, 10, was subjected to silyl protection at the hydroxy functionality, which was followed by reduction of the nitro group to afford 16 (Chart 3). A reaction sequence comprising protection of the amino group by Troc (17) and methylation at the nitrogen provided 18. Then, the carboxylic acid 15 and the amine 18 were connected by amide bond formation with 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT)19) as the coupling reagent, which was also used in the synthesis of Matsuda’s group.9–11)

Reagents and conditions: (a) In, AcOH, THF, 85°C, 90%; (b) TBSOTf, 2,6-lutidine, CH2Cl2, r.t., 76% (2 steps); (c) Troc-Cl, NaHCO3, THF–H2O (2 : 1), r.t., 98%; (d) MeI, NaH, DMF, 0°C, 90%; (e) Zn, NH4Cl, MeOH, r.t., 80%; (f) 15, DEPBT, NaHCO3, THF, r.t., 90%; (g) OsO4 (15 mol%), NMO, 1,4-dioxane–H2O (3 : 1), 60°C; (h) NaIO4, acetone, phosphate buffer (0.2 M, pH 7.4), r.t., 71% (2 steps); (i) H2 (1 atm), 5% Pd-C, MeOH, 60°C; (j) NaBH3CN, AcOH, AcOEt, r.t.; (k) (HCHO)n, NaBH3CN, r.t., 51%; (l) DMP, CH2Cl2, 0°C; (m) NaClO2, NaH2PO4, 2-methyl-2-butene, aq. THF, t-BuOH, r.t., 60% (2 steps); (n) 48% HF, CH3CN, 50°C, 45%.
The next step was oxidative cleavage of the olefin moiety of 19 to unveil the masked formyl group, followed by removal of the benzyloxycarbonyl (Cbz)- and benzyl-protecting groups under hydrogenolysis conditions (H2, 5% Pd-C). In this reaction, simultaneous cyclic imine formation took place to afford 21, although reduction of the C=N double bond was not observed, which was accomplished in a stepwise manner by treating NaBH3CN as the reductant. The resultant cyclic secondary amine structure was converted to the corresponding tertiary amine under reductive amination conditions with NaBH3CN. The sequence of Pinnick oxidation and the final deprotection achieved the total synthesis of (+)-caprazol.
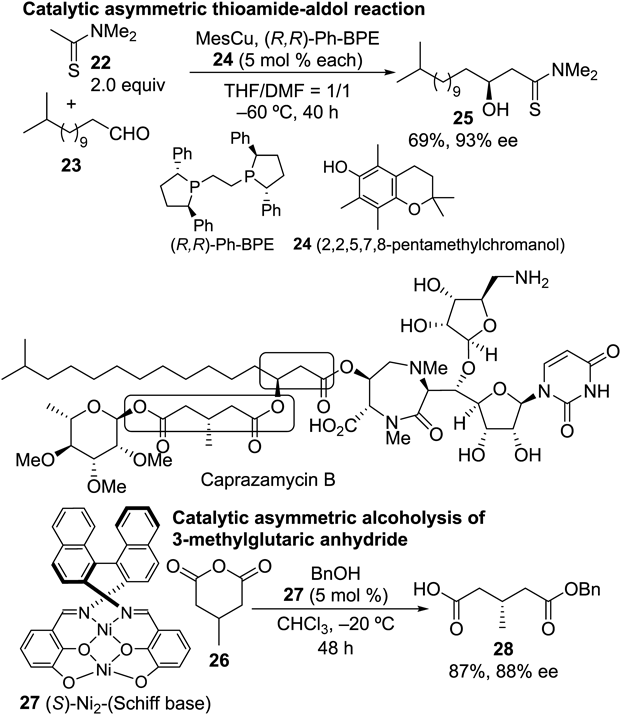
The subsequent task for the synthesis of caprazamycin B was preparation of the side chain moiety (western part) in a highly stereoselective manner.20) To address this issue, two catalytic asymmetric transformations were revealed to be effective, as shown in Chart 4. High enantioselectivity on the β-hydroxyester portion was achieved by the catalytic asymmetric thioamide-aldol reaction reported by Shibasaki’s group21–23): a catalytic amount of Cu(I) complexed with 1,2-bis(2,5-diphenylphospholano)ethane (Ph-BPE) promoted the addition of a thioamide 22 derived from acetic acid to an aldehyde 23 giving 25 in 93% ee. In the original report, no aldehyde having a long aliphatic carbon chain such as 23 was examined, meaning that the scope of the reaction was extended in this synthetic study. On the other hand, the unsymmetrically esterified glutaric acid found in this side chain portion was constructed by the newly developed catalytic asymmetric alcoholysis conditions.24) Toward this end, the binuclear nickel-chiral Schiff base complex 27, studied extensively by Shibasaki’s group, was the catalyst of choice.25–28) For this type of transformation, chiral organocatalysts including cinchona alkaloids have been widely used,29) although the availability of conditions for glutaric anhydride with sterically small substituents at the 3-position was limited. Moreover, naturally occurring alkaloids or their synthetic derivatives do not always guarantee the same level of selectivity upon obtaining enantiomeric products30–32) since pseudo-enantiomeric organocatalysts were used for this purpose. In this regard, metallic conditions can be used complementarily, and 88% ee of benzyl hemiester 28 was obtained with the use of 5 mol% of catalyst (Chart 4). That was one of the highest levels of selectivity observed for the 3-methylglutaric anhydride 26 at the time.
Subsequently, synthesis of the western part was straightforward, as seen in Chart 5. The aldol adduct 25 was easily converted to the corresponding aldehyde after protection of the hydroxy group as a tert-butyldiphenylsilyl (TBDPS) ether, which was followed by Pinnick oxidation, esterification, and desilylation to produce the β-hydroxyester 29. The free carboxyl portion of the hemiester 26 was condensed with the rhamnose derivative 30 to afford 31, which was subjected to coupling with 29 under Yamaguchi conditions followed by hydrogenolysis, giving rise to the western building block 32.
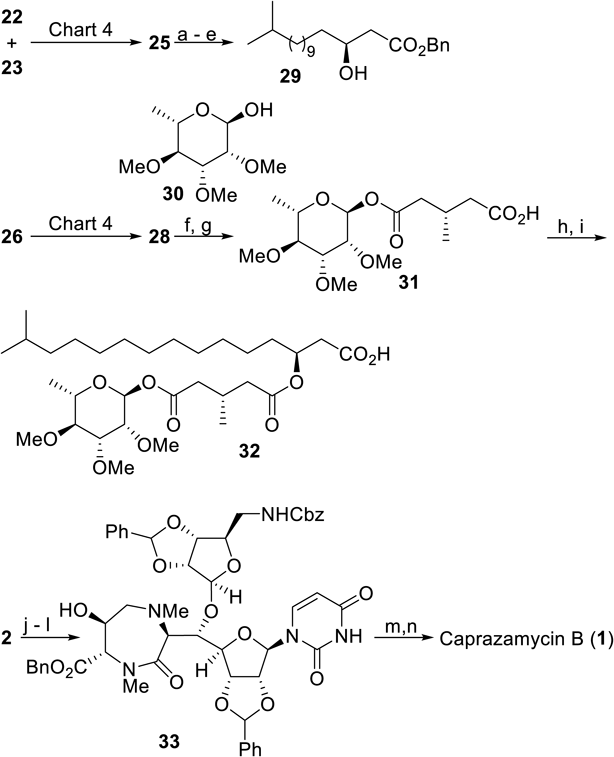
Reagents and conditions: (a) TBDPSOTf, 2,6-lutidine, CH2Cl2, 0°C, 95%; (b) MeOTf, Et2O, r.t., then LiAlH(Ot-Bu)3, Et2O, −30°C, 72%; (c) NaClO2, NaH2PO4, 2-methyl-2-butene, aq. THF, t-BuOH, r.t. to 0°C, 92%; (d) BnOH, DCC, DMAP, CH2Cl2, 0°C to r.t., 84%; (e) HF·pyridine, THF, 0°C to r.t., 80%; (f) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, 30, THF, r.t., 70%; (g) H2 (1 atm), 10% Pd-C, AcOEt, r.t., 98%; (h) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, 29, THF, r.t., 90%; (i) H2 (1 atm), 10% Pd-C, AcOEt, r.t., >99%; (j) NaBH3CN, AcOH, AcOEt, r.t.; (k) (HCHO)n, NaBH3CN, r.t., 51%; (l) DMP, CH2Cl2, 0°C; (m) NaClO2, NaH2PO4, 2-methyl-2-butene, aq. THF, t-BuOH, r.t., 60% (2 steps); (n) 48% HF, CH3CN, 50°C, 45%.
The subsequent coupling of 32 and the caprazol derivative 33 should have furnished all the requisite skeletal carbon atoms, although this proved troublesome owing to facile elimination of the β-hydroxyester substructure under most acylation conditions. Only Shiina’s protocol33) afforded the desired product in acceptable yield. Then, global deprotection in a manner similar to Takemoto’s route12) completed the synthesis of caprazamycin B.34) Later, catalytic asymmetric total synthesis of CPZEN-45 was also achieved,35) in which the aldol reaction to connect the uridine and diazepinone moieties was catalyzed by chiral linked-BINOL complexed with Zn(II) developed by Shibasaki and colleagues36–38) to improve the diastereoselectivity to as high as 79 : 1.
Ample evidence has accumulated that the proliferation of tumor cells in tumor tissues is under (at least partially) the control of signals emitted from the surrounding stromal cells consisting of normal cells such as fibroblasts.39–42) In efforts to propose novel strategies for exploratory research on anticancer leads, Kawada et al. established assay systems to screen for natural products enabling more potent growth inhibition of tumor cells in the presence of the corresponding stromal cells than in their absence.43,44) Hit compounds from this assay system can affect signal transduction involving stromal cells; considering the greater genetic stability of normal cells compared with tumor cells, such molecules are expected to be less prone to acquired resistance than molecular target anticancer agents. Figure 2 exemplifies a portion of hit compounds, which were actually synthesized.
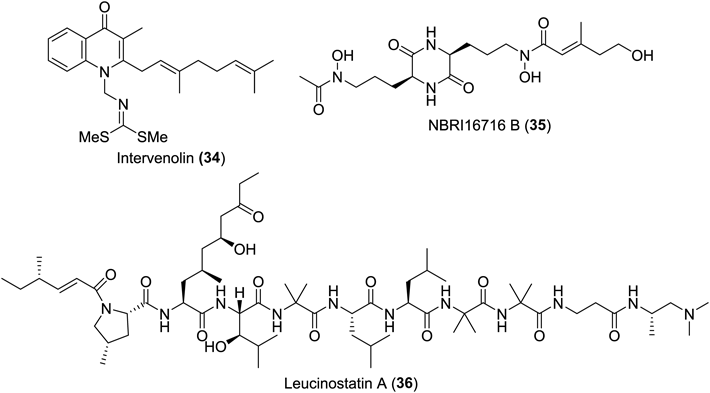
Intervenolin (34), produced by a strain of Nocardia, inhibits the growth of MKN-74 and MKN-7 gastric cancer cells co-cultured with Hs738 gastric stromal cells, and of Hs738 colorectal cancer cells co-cultured with CCD-18Co stromal cells.45) A diketopiperazine-based natural product, NBRI16716 B (35),46) and leucinostatin A (36)47) were found to decelerate the growth of DU-145 prostate cancer cells co-cultured with the corresponding PrSC stromal cells. The latter, a long-known natural product with a nonapeptide structure and diverse biological activities,48–57) reduced the expression of insulin-like growth factor I (IGF-I). However, at the outset of this research, the primary molecular targets of these three hit compounds were not known, which motivated my synthetic study aiming at the development of more potent analogues through SAR research and crafting molecular probes to bait counterpart proteins. This section focuses on efforts involving intervenolin (34) and leucinostatin A (36); a report on the synthetic study of NBRI16716 B (35) was also published elswhere.58)
Intervenolin has a chromophore of a 4-quinolone ring system substituted with two characteristic side chains, the iminodithiocarbonate substructure at the 1-position and the geranyl group at the 2-position, respectively. The former is rare even when looking into whole known chemical entities, and it is no surprise that this substructure was found in natural products for the first time. The latter also has not appeared frequently in the literature; the exception includes a report on structurally related anti-Helicobacter pylori natural products from the Pfizer team.59) The synthetic methods to install these side chains were unprecedented.
The synthesis of intervenolin has two key transformations, Suzuki–Miyaura coupling to introduce the geranyl group, and a thermal rearrangement from thiocyanate to isothiocyanate,60) as seen in Chart 6. To enable the cross coupling with geranylboronate 39, a quinoline derivative 38 was chosen as the substrate; the tert-butyldimethylsilyl (TBS) group was sequestered under the reaction conditions to afford 40 which has exactly the same structure as one of Pfizer’s compounds, CJ-13136. The substrate 38 was prepared by a combination of standard methods from 37. Deprotonation with an alkoxide bearing lithium as the countercation allowed selective introduction of the thiocyanate moiety at the 1-position.61–64) The presence of 41 in the media was transient with spontaneous rearrangement to the isothiocyanate 42. A one-pot protocol comprising the addition of methanethiolate and the subsequent methylation of the thiolate anion thus generated gave intervenolin.65) It is notable that the synthetic protocol is scalable: more than 1 g of intervenolin is easily obtained by a single run-through. An adequate amount of synthesized sample enabled in vivo examination of the antitumor effects in model mice inoculated with Hs738 cells, which clearly showed that intervenolin shrank tumor size even though the activity was not yet outstanding. Encouraged by this result, a primary SAR study was carried out66,67) (Chart 7).
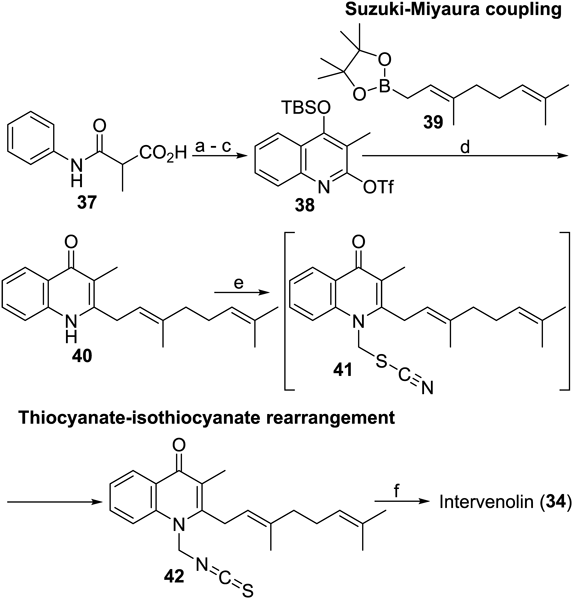
Reagents and conditions: (a) Eaton’s reagent, 80°C, 89%; (b) TBSCl, imidazole, DMF, r.t., 89%; (c) Tf2O, 2,6-lutidine, CH2Cl2, r.t., 92%; (d) 39, Pd(PPh3)2Cl2, 2 M NaHCO3 aq, EtOH–toluene (5 : 2), 90°C, 70%; (e) ClCH2SCN, LiOt-Bu, THF, r.t., 80%; (f) NaSMe, CH3CN, r.t., then MeI, CH3CN, r.t., 43%.
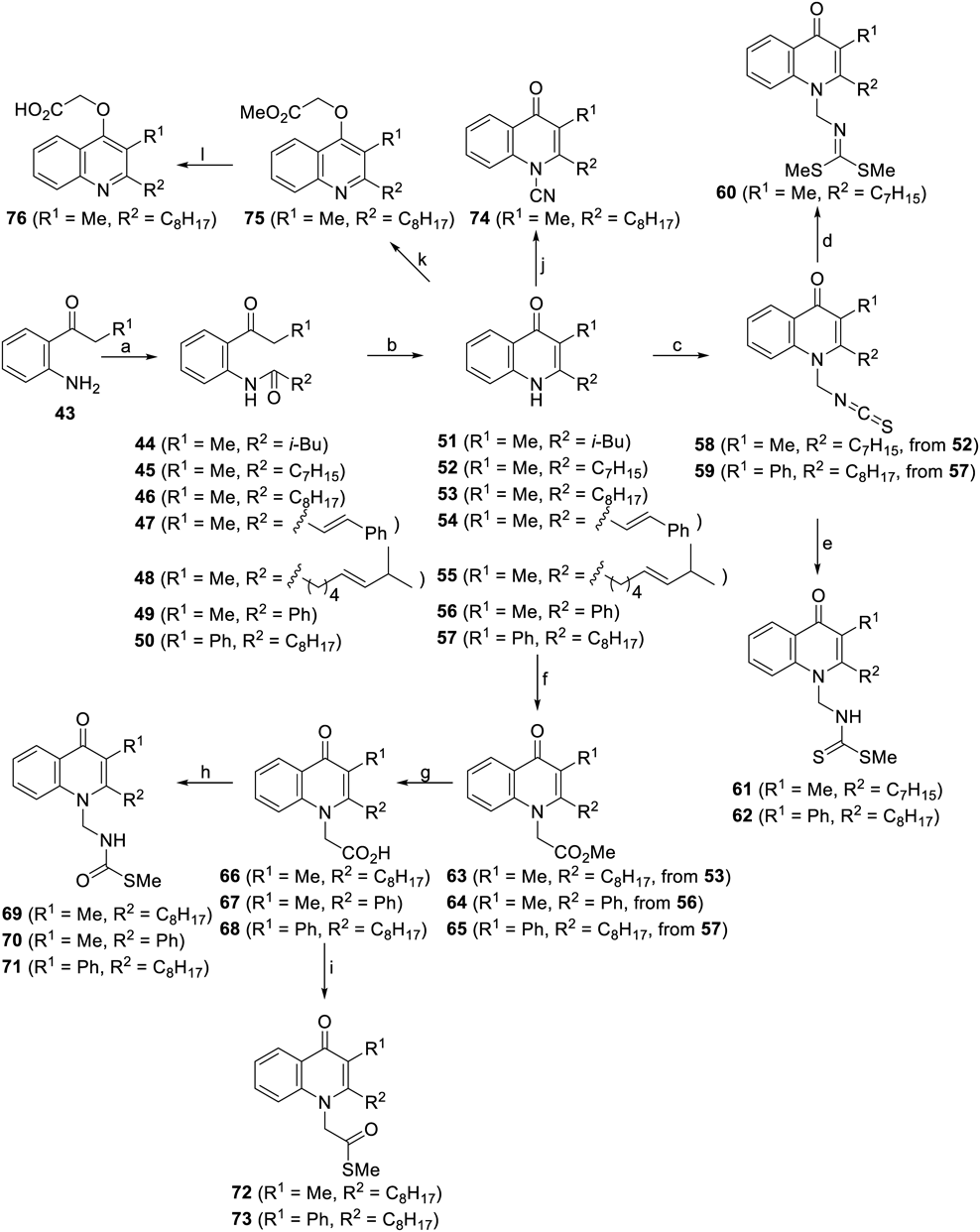
Reagents and conditions: (a) R2COCl, Et3N, CH2Cl2, r.t.; (b) NaOH, 1,4-dioxane, 110°C; (c) ClCH2SCN, LiOt-Bu, THF, r.t.; (d) NaSMe, CH3CN, r.t., then MeI, CH3CN, r.t.; (e) NaSMe, CH3CN, r.t.; (f) BrCH2CO2Me, LiOt-Bu, THF, 80°C; (g) 2 M NaOH, EtOH–THF, r.t.; (h) DPPA, Et3N, THF, 80°C, then NaSMe, 80°C; (i) NaSMe, DPPA, Et3N, THF, 80°C; (j) BrCN, LiOt-Bu, r.t.; (k) BrCH2CO2Me, K2CO3, DMF, 80°C; (l) 2 M NaOH, EtOH–THF, r.t.
The intermediates 51–57 were prepared starting from aniline derivative 43 by acylation to attach the side-chain portion at the 2-position (44–50) followed by dehydrative annulation. Installation of the 1-side chain was achieved by substitution with suitable electrophiles to give 58 and 59. The reaction sequence in Chart 7 from isothiocyanate 58 afforded 60 with the same pendant structure as that of intervenolin and a saturated long aliphatic chain instead of the geranyl part. Quenching was performed before methylation in this protocol for the formation of dithiocarbamate residues (61 and 62). Introduction of the acetate substructure at the 1-position was feasible to access 63–65, which were subsequently converted to carboxylic acid analogues 66–68 and then to thiocarbamates 69–71 and thioesters 72 and 73 in a divergent manner. On the other hand, changing the countercation to potassium upon the installation of an acetate unit gave the quinoline analogue 75 which was readily transformed into the carboxylic acid congener 76.61–64)
In Table 1, the antiproliferative activity of intervenolin analogues against MKN-74 cells from gastric cancer in the presence (co-cultured) or absence (monocultured) of the corresponding stromal cells (Hs738) together with acute toxicity in mice (in vivo maximum tolerated dose. MTD) is summarized. First, the effect of 2-substituents was examined using compounds without a pendant structure at the 1-position. Introduction of a long linear aliphatic chain (octyl, 53) kept the activity at almost the same level as that of 40 (geranyl) (0.011 vs. 0.010 µg/mL). The influence of the length is notable: an alkyl chain one carbon shorter decreased the IC50 value to 0.71 µg/mL (52). A short branched chain was not tolerable (51, 1.32 µg/mL), whereas methyl substituents and unsaturation at the periphery of the long alkyl group was allowed (55, 0.012 µg/mL). A styryl group at the 2-position completely disrupted the biological activity (54). For all the active analogues, selectivity between co-cultured and monocultured conditions were higher than those of intervenolin 34 and CJ-13,136 40, although substantial acute toxicity was unfortunately observed (1.56–25.0 mg/kg). This drawback was partly addressed by the introduction of a side chain at the 1-position.
| Compound | In vitro IC50 (µg/mL) | In vitro MTD (mg/kg) | |
|---|---|---|---|
| Cocultured | Monocultured | ||
| 34 | 0.17 | 3.0 | >50 |
| 40 | 0.010 | 0.25 | 2.5 |
| 51 | 1.32 | >100 | 25.0 |
| 52 | 0.71 | >100 | 6.25 |
| 53 | 0.011 | >100 | 12.5 |
| 54 | >100 | >100 | 12.5 |
| 55 | 0.012 | >100 | 1.56 |
| 57 | >10 | >10 | NT |
| 58 | 0.12 | 90.0 | 6.25 |
| 60 | 0.78 | 1.36 | >50 |
| 61 | 2.91 | 40.0 | >50 |
| 62 | 5.9 | ca. 10 | NT |
| 63 | 0.53 | >100 | 6.25 |
| 64 | >10 | >10 | >50 |
| 65 | 5.9 | >10 | >50 |
| 66 | 4.15 | 71.0 | >50 |
| 67 | >10 | >10 | NT |
| 68 | >10 | >10 | >50 |
| 69 | 0.27 | 5.84 | >50 |
| 70 | >10 | >10 | >50 |
| 71 | 4.3 | 9.3 | >50 |
| 72 | 1.40 | 5.84 | >50 |
| 73 | >10 | >10 | NT |
| 74 | 0.007 | >100 | 6.25 |
| 75 | 0.35 | 28.1 | >50 |
| 76 | 0.37 | 2.95 | >50 |
Isothiocyanate (58), acetate ester (63), and cyano (74) functionalities still showed higher toxicity, although 74 exhibited potent antiproliferative activity (0.007 µg/mL). The thiocarbamate analogue 69 displayed moderate growth-inhibitory activity in a selective manner under co-cultured conditions (0.27 µg/mL), and no severe toxicity was observed. Installation of dithiocarbamate (61), acetate (66), and thiol ester (72) functionalities reduced the activity (1.40–4.15 µg/mL). An iminodithiocarbonate substructure (61) was preferable in terms of toxicity, whereas selectivity between co-cultured and monocultured conditions was diminished. A phenyl substituent at the 2- or 3-position in general reduced the antiproliferative activity. Surprisingly, analogues with quinoline skeletons (75 and 76) also inhibited the growth of the tumor cells under co-cultured conditions (0.35 and 0.37 µg/mL, respectively) without acute toxicity in mice. This class of analogues can be recognized as backup compounds for anticancer leads. A convincing mode of action for the antiproliferative activity is currently being clarified, which will be reported in the near future.
The core structure of intervenolin, 4-quinolone, is widely known as a chromophore of antibiotics including CJ-13,136 (40), which led us to investigate the antibacterial activity of the analogues described above, specifically their anti-H. pylori activity. Table 2 shows the results against three strains of H. pylori, and pathogenic or nonpathogenic species of bacteria compared with clarithromycin, a clinically used anti-H. pylori agent. The anti-H. pylori activity of intervenolin (34), 52, 53, 55, and 58 was generally comparable to that of the positive control, clarithromycin (MIC ranging from 0.0078 to 0.0156 µg/mL), which was substantially more potent than the benchmark compound CJ-13,136 (40, 0.25 and 0.5 µg/mL). Notably, the selectivity of the anti-H. pylori activity of these compounds over other species was outstanding: 52, 53, and 55 showed essentially no antibacterial activity toward Staphylococcus aureus, Enterococcus faecalis, and Escherichia coli. No obvious relationship was seen between the antiproliferative activity and antibacterial activity of the tested compounds.
| Test organisms compound | Helicobacter pylori JCM 12093 | H. pylori JCM 12095 | Staphylococcus aureus FDA209P | Enterococcus faecalis JCM5803 | Escherichia coli K-12 |
|---|---|---|---|---|---|
| 34 | 0.0156 | 0.0078 | 64 | >128 | >128 |
| 40 | 0.5 | 0.25 | 128 | >128 | 128 |
| 52 | 0.0156 | 0.0156 | >128 | >128 | >128 |
| 53 | 0.0078 | 0.0078 | >128 | >128 | >128 |
| 54 | 1 | 0.5 | >128 | >128 | >128 |
| 55 | 0.0078 | 0.0156 | >128 | >128 | >128 |
| 58 | 0.0156 | 0.0156 | 4 | >128 | >128 |
| 60 | 2 | 2 | 4 | >128 | >128 |
| 61 | 0.0312 | 0.0625 | >128 | >128 | >128 |
| 63 | 2 | 2 | >128 | >128 | >128 |
| 66 | 1 | 0.25 | 128 | >128 | 128 |
| 64 | 0.25 | 0.5 | 128 | >128 | 128 |
| 65 | 4 | 8 | 4 | >128 | 128 |
| 76 | 1 | 0.5 | >128 | >128 | 128 |
| CAM | 0.0078 | 0.0078 | <0.125 | 0.5 | 16 |
CAM: clarithromycin.
Although it is beyond the scope of this review article, the biological background of this intriguing anti-H. pylori activity is worth mentioning.68) Clinically, triple combination therapy with clarithromycin, amoxicillin (antibiotics), and a proton pump inhibitor such as omeprazole is currently the first choice to eradicate H. pylori in gastrin. Although 61 exhibited slightly less antibacterial activity (0.0312 and 0.0625 µg/mL) than 53 and clarithromycin, it showed superior therapeutic efficacy to the triple-therapy cocktail using an in vivo mouse model. In part, 61 reduces the colony-forming unit count of H. pylori by inhibition of dihydroorotate dehydrogenase (DHODH), which is one of the essential enzymes for H. pylori. The lack of a salvage pathway for pyrimidine biosynthesis necessitates DHODH within the de novo pathway, which makes inhibition of this enzyme lethal. Another mechanism is also operative: 61 suppresses the function of urease of H. pylori that degrades urea to quench the acidic environment by the production of ammonia from urea. The compromised activity level of ureases leads to H. pylori death by acid in gastrin.
Another synthetic target, leucinostatin A, is composed of leucin and five types of unusual amino acids, β-alanine, 2-aminoisobutyric acid (Aib), threo-β-hydroxyleucine (HyLeu), 2-amino-6-hydroxy-4-methyl-8-oxodecanoic acid (AHMOD), and 4-methylproline (MePro). Chart 8 displays the catalytic asymmetric nitroaldol reaction with LLB* (79) developed by Shibasaki, Sasai and colleagues,69,70) which was utilized in this study to install all the carbon framework with the correct configuration. In the original report, 2-nitroethanol 77 was found to be one of the best substrates to attain perfect syn-selectivity and almost perfect enantioselectivity, whereas an α-branched aldehyde such as 78 was not examined. In the author’s study, the nitroaldol adduct 80 was obtained with essentially the same stereoselectivity (dr = 12 : 1, 96% ee), although increased catalyst loading up to 9 mol% was required.
With the intermediate 80 in hand, the 9-fluorenylmethyloxycarbonyl (Fmoc)-protected HyLeu derivative 83 was synthesized by a sequence of standard manipulation of functional groups and protecting groups (Chart 9): reduction of the nitro group followed by Fmoc protection gave 81, which was subjected to silylation at the secondary hydroxy group leading to 82 and Pinnick oxidation.
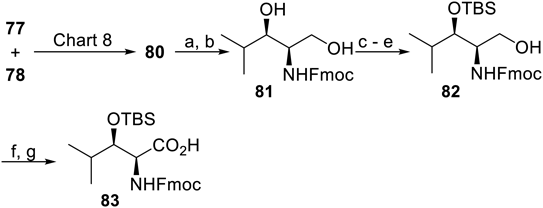
Reagents and conditions: (a) H2, 10% Pd-C, MeOH, r.t.; (b) Fmoc-Cl, Na2CO3, aq. THF, 0°C, 97% (2 steps); (c) TESOTf, 2,6-lutidine, CH2Cl2, r.t., 97%; (d) TBSOTf, 2,6-lutidine, CH2Cl2, r.t., 86%; (e) PPTS, MeOH, r.t., 99%; (f) DMP, NaHCO3, CH2Cl2, r.t.; (g) NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH–H2O–THF (3 : 5 : 2), r.t., 88% (2 steps).
Another amino acid residue, AHMOD, showing the most complicated structural features including three stereocenters was prepared starting from an array of reactions, as shown in Chart 10. The methyl group at the 4-position was furnished by taking advantage of the catalytic asymmetric desymmetrization reaction demonstrated in Chart 4.24) A reduced amount of catalyst (from 5 to 2.5 mol%) and methanol instead of benzyl alcohol as the nucleophile allowed the reaction to proceed with the same degree of selectivity. According to the reported protocol, a hemiester 84 was transformed into aldehyde 85.71) Subsequent conversion of aldehyde to the diphenylidene derivative 86 was indispensable to achieve a reasonable level of the diastereomeric ratio in the second key stereoselective transformation, and a Strecker-type reaction was required for the correct configuration on the α-carbon of this amino acid unit.72,73) Adopting a two-step procedure, a substrate for this reaction having a 9-fluorenylimine structure (87) was accessible.

Reagents and conditions: (a) diethyl diphenylmethylphosphonate, LHMDS, THF, 0°C, 60%; (b) DIBAL-H, CH2Cl2, −78°C, 90%; (c) 9-fluorenylamine, toluene, 0°C, >99%; (d) DDQ, THF, 0°C, then 1 M HCl, THF, r.t., 68%; (e) 4 M HCl, 1,4-dioxane, MeOH, 50°C, 91%; (f) Boc2O, Na2CO3, aq. THF, r.t., 95%; (g) Boc2O, DMAP, CH3CN, r.t., 90%; (h) O3, pyridine, CH2Cl2, −78°C, 80%.
In the present case, 9 mol% of the catalyst 88, a Lewis acidic Al(III) complexed with a BINOL-based chiral framework with Lewis basic phosphine oxide moieties, was required for the reaction to proceed at an acceptable rate. Other than the cyan source trimethylsilyl cyanide and an additive phenol, H2O was also needed although its role is not yet clear. As mentioned above, the bulkiness of the remote part of the substrate influences the diastereoselectivity (5.5 : 1); TBS ether instead of the diphenylidene resulted in a substantial loss of selectivity (1.3 : 1). Then the cyano group was transformed into methyl ester by exposure to acidic media containing methanol. Subsequently, the protecting group for amine was changed to afford 90, which was followed by unveiling the masked aldehyde by ozonolysis to give 91.
For the secondary alcohol part, aldol-type reactions were screened (failed examples included LLB-catalyzed conditions employing 2-propanone as a counterpart). As a result, the catalytic thioamide-aldol protocol described in the synthesis of caprazol21–23) was successful. The thioamide 92 and aldehyde 91 were applied to the protocol shown in Chart 11 with (S,S)-Ph-BPE as the chiral ligand to produce 93 with perfect diastereoselectivity. The configuration of the newly generated secondary alcohol was confirmed to be S by derivatization to a known molecule. After protection of the revealed hydroxy functionality as a TBS ether (94), the thioamide moiety was converted to ethyl ketone 95. Formation of a methylthioiminium intermediate with MeOTf was followed by treatment with EtMgBr and subsequent hydrolysis by weak acid (diluted hydrochloric acid in tetrahydrofuran (THF) or NH4Cl). This was the first case of converting the thioamide adduct to a simple ketone other than a methyl ketone in a one-pot procedure. The following hydrolysis of ester under standard basic or acidic conditions suffered from β-elimination of the siloxy moiety. Only Me3SnOH provided the desired carboxylic acid 96 in good yield.74)
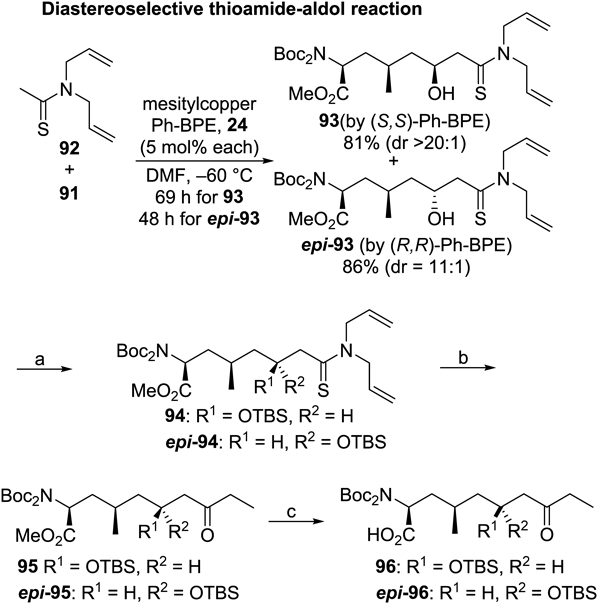
Reagents and conditions: (a) TBSOTf, 2,6-lutidine, CH2Cl2, r.t., 94% from 93, 83% from epi-93; (b) MeOTf, CH2Cl2, 40°C, then EtMgBr, THF, −78°C, then 1 M HCl, aq. THF, 0°C, 44% from 94, MeOTf, CH2Cl2, 40°C, then EtMgBr, THF, −78°C, then NH4Cl, aq. THF, 0°C, 56% from epi-94; (c) Me3SnOH, 1,2-dichloroethane, 120°C or 110°C, 82% from 95, 99% from epi-95.
The final stage of the total synthesis (Chart 12) was elongation of the peptide from 97 which was prepared on resin. The terminal tertiary amine unit 98,75) and the acylated MePro derivative 10076) could be readily obtained according to the procedures in the literature. Lower accessibility of amino acid units 83 and 96 is the reason for liquid phase synthesis after 97. The global deprotection of 99 was accomplished by treatment with Lewis acid, BF3·OEt2,77) where I believed that the total synthesis of leucinostatin A (46) was successful78) judging from the good agreement of the 1H- and 13C-NMR data of synthetic and natural samples. However, the retention time of HPLC of each sample was slightly different even after thorough and repeated purification, and the antiproliferative activity of the synthetic material against DU-145 cells in the presence of PrSC was around two-fold weaker than that from the natural one (IC50; natural, 0.029 µg/mL; synthetic, 0.058 µg/mL). At that time, I suspected that the structure in the literature was reported with incorrect assignment of the spectral data. Brimble and colleagues pointed out that the absolute configuration of the 6-hydroxy group of AHMOD within two peptaibol natural products, culicinin D79) and trichodermin A,80) should be R, not S. The conclusion was drawn based on the comparison of the NMR data of each synthesized diastereomer and the reported values, although the data from the former were not completely coincident with those from the latter. The conclusion relied mainly on the spectral similarity, and biological testing was not feasible, leaving overall ambiguity on the new stereochemical assignment. Although the general reliability of determination of the absolute configuration by crystal structure (as is the case for leucinostatin A)81) partly allayed the suspicion of misassignment, synthesis of the R-isomer was undertaken for strict comparison of the spectral and biological data (Charts 11 and 12).
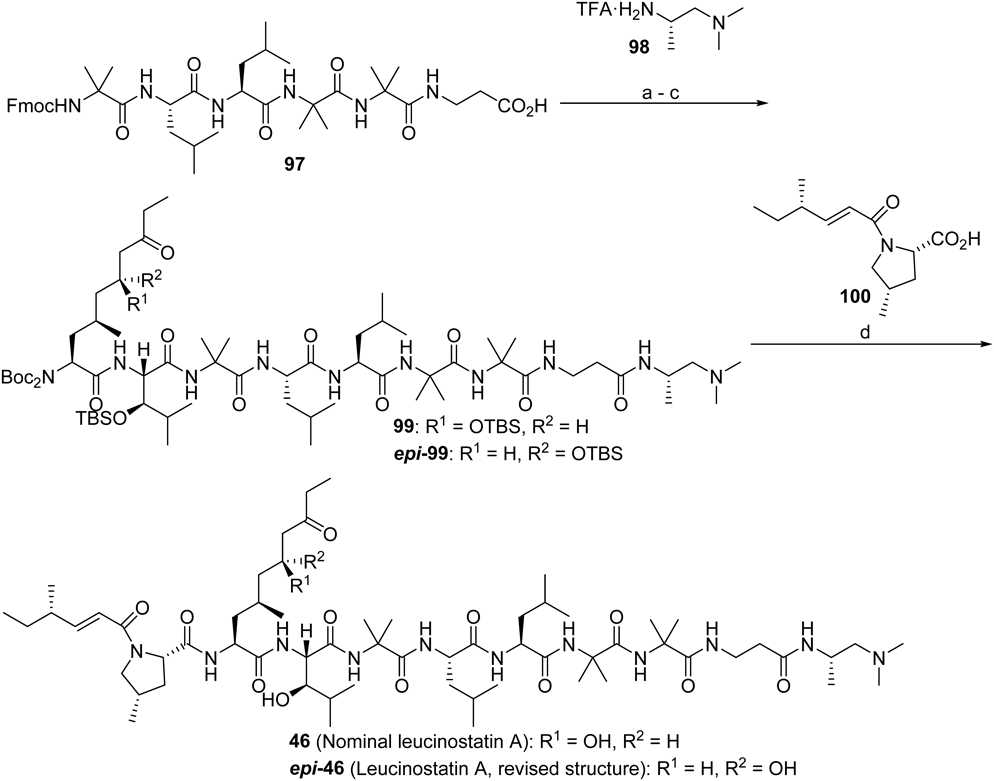
Reagents and conditions: (a) 98, HATU, DIPEA, DMF, r.t., 62%; (b) 20% piperidine, DMF, r.t., then 83, HATU, DIPEA, DMF, r.t., 76%; (c) 20% piperidine, DMF, 23°C, then 96 or epi-96, HATU, DIPEA, DMF, r.t., 56% with 96, 46% with epi-96; (d) BF3·OEt, MS 4A, CH2Cl2, 0°C to r.t.; 100, HATU, DIPEA, DMF, r.t., 44% from 99, 32% from epi-99 (2 steps).
The synthetic pathway was essentially the same as for 46 and although a mismatch of stereochemistry between 91 and the catalyst decreased the diastereoselectivity slightly, the 11 : 1 ratio still gave rise to epi-93. Surprisingly, all the NMR data from the synthetic (epi-46) and natural materials were once again indistinguishable. Moreover, growth inhibition of DU-145 under co-cultured conditions exerted by both the samples were exactly the same (IC50; natural, 0.029 µg/mL; synthetic epi-46, 0.029 µg/mL), and HPLC profiles were also identical. Therefore, the absolute configuration of the 6-position of AHMOD in leucinostatin A was revised to be R.78)
Recently, a SAR study of leucinostatin A utilizing the present synthetic route has been carried out to reveal that the whole length of the peptide chain is important, but not for the side chain structure of AHMOD.78,82) So far, the primary molecular target of leucinostatin A in terms of intervention in tumor–stroma interactions has not been clarified. The design and synthesis of molecular probes to address this issue are currently underway.
The establishment of synthetic pathways to biologically active natural products broadens the scope of these molecules as candidates for lead compounds of clinically useful drugs by generating more potent, pharmacologically relevant analogues. The identification of primary molecular targets of hit molecules discovered by phenotypic screening can provide novel guidelines for exploratory research on medical seeds. With this background in mind, the author carried out synthetic studies on biologically active natural products and their related compounds. In this review, studies on leptolyngbyolides (cytotoxic marine macrolides),83,84) BE-54017 (V-ATPase inhibitor),85) tyropeptin (proteasome inhibitor),86–89) androprostamine A and resormycin (antiproliferative agents against prostate cancer cells)90) are not described, although the details can be found in the publications indicated. Synthetic chemistry and natural products have long been friends and provided many opportunities to create medicines which in turn improved public health. Even scarcity of occurrence from natural resources and complex structures could be overcome with the immense power of synthetic methods accumulated throughout the history of science. It should be mentioned that natural products in a conventional sense no longer occupy an inviolable position among medical leads. Nevertheless, it remains true that the occasionally discovered novel chemical entity from nature could take us beyond the horizon where unmet medical needs are waiting for these molecules to be utilized.
The author appreciates continuous support from Prof. Dr. Masakatsu Shibasaki throughout this research. I am grateful to Dr. Manabu Kawada, Dr. Hikaru Abe, Dr. Purushothaman Gopinath, Dr. Masayuki Igarashi, Dr. Tomokazu Ohishi, Dr. Junjiro Yoshida, Dr. Lu Wang, Dr. Hitoshi Ouchi, Dr. Gandamala Ravi, Dr. Takashi Masuda, Dr. Jin Cui, Dr. Yoji Umezawa, Dr. Isao Momose, Dr. Toshifumi Takeuchi, Dr. Tomoyuki Kimura, Prof. Dr. Masaya Imoto, Prof. Dr. Kiyotake Suenaga, Prof. Dr. Toshiaki Teruya, Dr. Osamu Ohno, Dr. Maho Morita, Ms. Chiharu Sakashita, Mr. Shun-ichi Ohba, Mr. Hiroyuki Inoue, Mr. Tohru Masuda, and Ms. Chigusa Hayashi for fruitful collaboration. I would like to thank Prof. Dr. Akio Nomoto, Dr. Yuzuru Akamatsu, Dr. Daishiro Ikeda, and Dr. Yoshikazu Takahashi for their encouragement. I appreciate Dr. Naoya Kumagai, Dr. Toshiaki Miyake, Dr. Yoshiaki Takahashi, Dr. Yoshimasa Ishizaki, Dr. Minoru Yonezawa, and Ms. Maya Umekita for valuable suggestions and discussions. I thank Dr. Ryuichi Sawa, Dr. Kiyoko Iijima, Ms. Yumiko Kubota, and Ms. Yuko Takahashi for the collection of spectral data. The author is grateful to the JST (ACT-C), Japan Society for the Promotion of Science (JSPS) (KAKENHI, Grant-in-Aid for JSPS fellow), AMED (P-CREATE), the Novartis Foundation, Sumitomo Foundation, Uehara Memorial Foundation, Takeda Science Foundation, Astellas Foundation for Research on Metabolic Disorders, and Suzuken Memorial Foundation for their financial support.
The author declares no conflict of interest.