Notes
Synthesis, Anticancer Activity and Molecular Modeling Studies of Novel Chalcone Derivatives Containing Indole and Naphthalene Moieties as Tubulin Polymerization Inhibitors
2019 Volume 67 Issue 7 Pages 725-728

Details
2019 Volume 67 Issue 7 Pages 725-728
Eighteen novel chalcone derivatives containing indole and naphthalene moieties (2–19) were synthesized and characterized by 1H-NMR, 13C-NMR and high resolution (HR)-MS spectra. All compounds were evaluated for their in vitro cytotoxic potential against human hepatocellular carcinoma (HepG2), human colon carcinoma (HCT116) and human breast adenocarcinoma (MCF-7) cell lines. Among them, compound 2, 3, 4 and 7 showed potent activities against tested cancer cell lines. More significantly, compound 7 exhibited the most potent cytotoxic activity against HepG2, HCT116 and MCF-7 with IC50 values of 0.65, 1.13 and 0.82 µM, respectively. Furthermore, flow cytometry analysis indicated that compound 7 arrested cancer cells in G2/M phase. The compound 7 also displayed significant inhibition of tubulin polymerization (IC50 = 3.9 µM). Finally, molecular docking studies were performed to explore the possible interactions between compound 7 and tubulin binding pockets.
Microtubule, the major component of cytoskeleton, is considered to play an important role in the number cellular processes including cell division, cell signaling, cell secretion, cell motility and intracellular transports.1,2) Therefore, the disruption of microtubule function will result in mitotic blockage and leading to tumor cell death by apoptosis.3) A number of antimitotic agents, such as the vinca alakaloids, taxanes, epothilone, and nocodazole, have been in clinical use for many years.4,5) Thus, discovery and development of new antimitotic agents for the treatment of cancer is an important research area in medicinal chemistry.
Chalcone is an important scaffold found in many naturally occurring and synthetic compounds, which displayed many pharmacological activities. During the recent decades, diverse chalcone derivatives displayed potent anticancer activity by inhibit tubulin polymerization or depolymerization.6–8) Indoles are very commonly skeleton in natural products and synthetic compounds, which have attracted great attention in medicinal chemistry over the past decade.9,10) It is important to point out that indole moiety is an important pharmacophoric fragment for design and development antitubulin agents as reported in the literature.11,12) Based on this, a large number of natural products and synthetic compounds containing indole moiety have been discovered as tubulin inhibitors for the treatment of cancer, and some of them have been approved for clinical use or entered clinical trials13–15) (Fig. 1). On the other hand, several compounds containing naphthalene moiety have been reported as potent anticancer agents that inhibit tubulin polymerization16–18) (Fig. 1).

Because of these observations and following our interest in design and synthesis of novel tubulin polymerization inhibitors,17,18) herein we report for the first time the synthesis of a novel series of chalcone derivatives containing indole and naphthalene moieties.
The synthesis of chalcone derivatives containing indole and naphthalene moieties (2–19) is shown in Chart 1. The compound 1 was prepared by Friedel–Crafts reaction. Claisen Schmidt condensation of aromatic ketone compound 1 with various commercial available indole aldehydes in the presence of KOH in methanol to afford the corresponding chalcone derivatives 2–5 in moderate yields. A series of indole chalcone derivatives with N-substitution 6–19 were prepared by reaction of compound 3 with various alkyl halides under basic conditions. To the best of our knowledge, all compounds (2–19) are new and have not been reported in the literature. Their chemical structures were characterized by 1H-NMR, 13C-NMR and high resolution (HR)-MS.

The anticancer activity of these new chalcones was tested by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay against human hepatocellular carcinoma (HepG2), human colon carcinoma (HCT116) and human breast adenocarcinoma (MCF-7). Cisplatin was used as positive control. The values of IC50 greater than 10.0 µM are considered inactive. The results were shown in Table 1. To the study of structure–activity relationship (SAR) of this class of compounds, we first synthesized compound 2–5 and study the effect of the position of the indole on anticancer activity. The results were shown that all of the tested compounds exhibited potent anticancer activity against tested cell lines. Among them, compound 3 with α,β-unsaturated ketone located at the C-5 position of the indole ring was found to be the most active compound. Shifting α,β-unsaturated ketone group to the other positions resulted in slight to moderate decreased anticancer activity. Since C-5 position of the indole ring is proved the most potent position, we then synthesized a series of indole chalcone derivatives 6–19 with N-substitution by reaction of compound 3 with various alkyl halides and evaluated the effect of the substitution at the N-1 position of indole ring. As shown in Table 1, introduction methyl group at the N-1 position result in slight increase of anticancer activity (7). However, the replacement of H with substitute benzyl group (6, 12–19) or other bulk alkyl group (8–11) at N-1 position of indole ring resulted in a remarkable increase of the biological activity.
| Code | Substitute position | R | IC50 (µM) | ||
|---|---|---|---|---|---|
| HepG2 | HCT116 | MCF-7 | |||
| 2 | 4 | H | 0.86 ± 0.12 | 6.61 ± 0.53 | 1.52 ± 0.09 |
| 3 | 5 | H | 0.74 ± 0.08 | 4.16 ± 0.32 | 1.21 ± 0.17 |
| 4 | 6 | H | 0.89 ± 0.07 | >10.0 | 2.02 ± 0.14 |
| 5 | 7 | H | 7.65 ± 0.33 | 9.11 ± 0.64 | 8.50 ± 0.48 |
| 6 | 5 | 4-(tert-Butyl)benzyl | >10.0 | >10.0 | >10.0 |
| 7 | 5 | Methyl | 0.65 ± 0.06 | 1.13 ± 0.08 | 0.82 ± 0.11 |
| 8 | 5 | Propyl | >10.0 | >10.0 | >10.0 |
| 9 | 5 | Isopropyl | >10.0 | >10.0 | >10.0 |
| 10 | 5 | Butyl | >10.0 | >10.0 | >10.0 |
| 11 | 5 | Isobutyl | >10.0 | >10.0 | >10.0 |
| 12 | 5 | Benzyl | >10.0 | >10.0 | >10.0 |
| 13 | 5 | 4-Fluorobenzyl | >10.0 | >10.0 | >10.0 |
| 14 | 5 | 4-Chlorobenzyl | >10.0 | >10.0 | >10.0 |
| 15 | 5 | 4-Bromobenzyl | >10.0 | >10.0 | >10.0 |
| 16 | 5 | 2-Fluorobenzyl | >10.0 | >10.0 | >10.0 |
| 17 | 5 | 3-Fluorobenzyl | >10.0 | >10.0 | >10.0 |
| 18 | 5 | 2,4-Dichlorobenzyl | >10.0 | >10.0 | >10.0 |
| 19 | 5 | 2-Bromobenzyl | >10.0 | >10.0 | >10.0 |
| Cisplatin | 4.95 ± 0.41 | 8.27 ± 1.58 | >10.0 | ||
In order to investigate whether the anticancer activities of this class of compounds by inhibiting tubulin polymerization, compound 7 was assessed for its inhibition of tubulin polymerization in vitro. Colchicine was used as a reference, while dimethyl sulfoxide (DMSO) was used as a vehicle control. Purified and unpolymerized tubulin was incubated with compound 7 or colchicine at the concentrations of 0.8, 1.5, 3.0, 6.0, 12.5 and 25.0 µM, respectively, and measured the change of absorbance at 350 nm. This result was indicated that compound 7 is a novel tubulin polymerization inhibitor, and showed a trend similar to colchicine. The IC50 of compound 7 and colchicine were 3.9 and 13 µM, respectively.
Flow Cytometric AssayTo examine the effect of compound 7 on the cell cycle, HepG2 cells were treated with compound 7 at dose of 1.0 or 2.0 µM for 24 h and subjected to flow cytometric analysis. The data obtained clearly indicate that compound 7 cause cell-cycle arrest in G2/M in a dose-dependent manner as comparison with untreated control (Fig. 2). At 1.0 µM, compound 7 showed 79.63% cell accumulation in G2/M phase, whereas at 2.0 µM it exhibited 92.38% cell accumulation in G2/M phase.

(A) control; (B) compound 7 (1.0 µM); (C) compound 7 (2.0 µM).
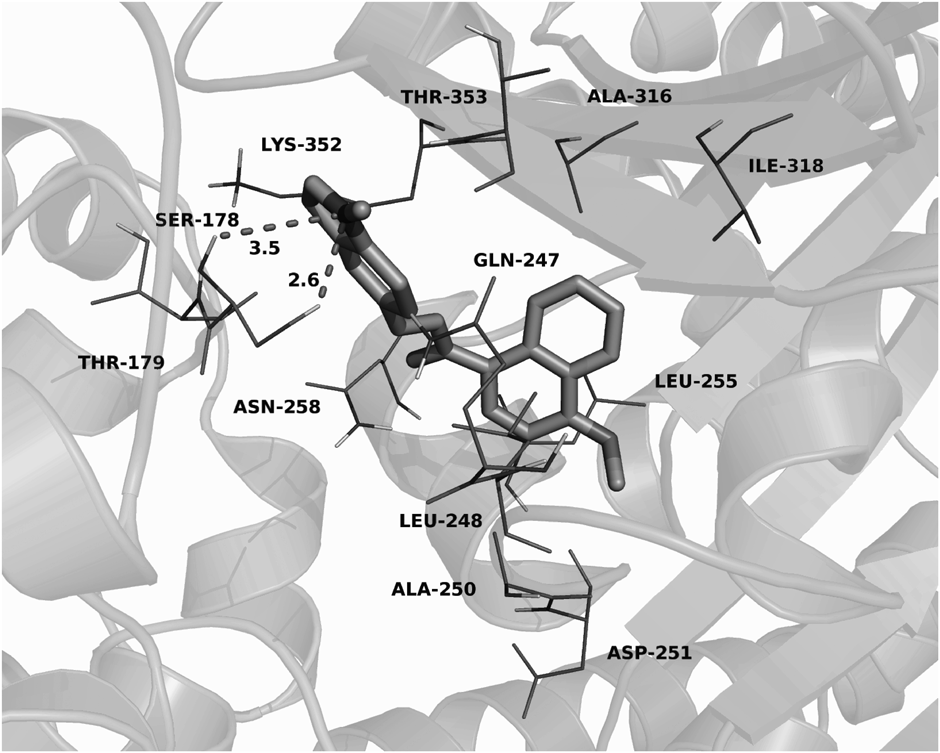
To clarify the binding mode of these novel chalcones, compound 7 was docked into the active sites of tubulin by using Autodock VINA 1.1.2.19) The result was shown in Fig. 3. The compact conformation of compound 7 is the best docked poses, which has the lowest binding energy. The docking results revealed that 4-methoxynaphthyl group of compound 7 was bound into the hydrophobic pocket of tubulin and showing strong hydrophobic interactions with the residues leucine (Leu)-248, alanine (Ala)-250, Leu-255, Ala-316 and isoleucine (Ile)-318. The residue lysine (Lys)-352 is involved in cation-π interaction with the indole group of compound 7. The residue serine (Ser)-178 formed two hydrogen bond interactions (bond length: 2.6 and 3.5 Å) with the N-Me group of compound 7.
In conclusion, a new class of chalcone derivatives containing indole and naphthalene moieties were synthesized and screened for anticancer activity against several cancer cell lines. Compound 7 exhibited most potent cytotoxic activity against HepG2, HCT116 and MCF-7 with IC50 values of 0.65 ± 0.06, 1.13 ± 0.08 and 0.82 ± 0.11 µM, respectively. Mechanism study demonstrated that compound 7 could arrest cell-cycle progression and inhibit tubulin polymerization with IC50 value of 3.9 µM. Finally, molecular docking study revealed that compound 7 have hydrophobic, cation–π and hydrogen bonding interactions with the colchicine binding site of tubulin. Further studies on structural optimization and biological activities about this class of compounds are still in progress.
This work was supported by National Natural Science Foundation of China (81660574), one thousand Talents Program of Guizhou Province (the fifth group), basic research plan of Guizhou province ([2019]1256) and the Doctor Foundation of Guizhou Medical University ([2018]004).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.