Abstract
The 4-vinylpyrimidin-2-one nucleoside (T-vinyl) forms a cross-link with the RNA containing uracil at the complementary site at a high reaction rate. To obtain the stable T-vinyl derivative so that its reactivity is protected until it access to the target site, several derivatives were investigated, and the 2-thiopyridinyl- and 2-thiopyrimidinyl T-vinyl derivatives were determined to be good candidates. The 2-thiopyrimidinyl T-vinyl derivative was found to more efficiently cross-link with mRNA albeit having a better stability than the 2-thiopyridinyl T-vinyl derivative. The investigation using the luciferase (Luc) mRNA, the synthetic mRNA and non-cellular translation system revealed that the translation is terminated at the end of the cross-linked duplex between the mRNA and the oligoribonucleotide (ORN). Thus, the 2-thiopyrimidinyl T-vinyl derivative has successfully demonstrated both a good stability and high efficiency for the cross-linking reaction, and expanded its applicability in biological applications.
Introduction
The antisense oligonucleotide is required to form a stable double-stranded complex with the target RNA to function as in the regulation of splicing of pre-mRNA, translation of mRNA, and inhibition of microRNA (miRNA).1–3) For this purpose, a variety of nucleic acid analogs has been developed, which include locked nucleic acid (LNA), morpholino nucleic acid, peptide nucleic acid (PNA), etc. Cross-linking between the oligonucleotide and the target RNA is an alternative strategy to stabilize the double-stranded complex. For the intracellular application, the crosslinking oligonucleotide (ON) needs to satisfy (1) stability, (2) high reactivity and (3) high selectivity under the physiological conditions. Thus, cross-linking reagents with an inducible reactivity under certain specific conditions have been developed. Psoralen is activated by light irradiation that causes photo [2 + 2] cyclization with thymine and uracil to form a cyclobutane ring, and psoralen-conjugated antisense oligodeoxynucleotides (ODNs) were applied to inhibit the transcription and cell proliferation.4–7) The 3-cyanovinyl carbazole nucleoside (CNVK) exhibited ultrafast photo-cycloaddition to pyrimidine bases upon the irradiation of 366 nm light.8) Interestingly, the cyclobutane ring formation with cytosine induced deamination of the 4-amino group of cytosine.9) The furan ring was chemically oxidized to the 4-oxo-enal derivative, which was shown to form cross-links with adenine and cytosine at complementary positions of the target DNA in a sequence-dependent manner.10) The quinone methide unit was also applied to the DNA alkylation.11) The 2-amino-6-vinylpurine (2-AVP) is a nucleoside derivative, of which the vinyl group selectively reacts with the 4-amino group of the target cytosine. This crosslinking ODN was protected with a sulfide group and applied for inhibiting the protein expression in cells.12–14)
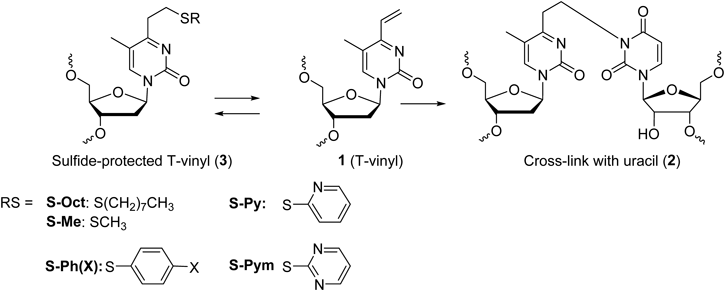
The 4-vinylpyrimidin-2-one nucleosides (T-vinyl, 1) exhibit an efficient and selective cross-link formation with the uracil base at the complementary site in the RNA, producing more than a 90% yield for a 15-min reaction time.15) For biological application, the cross-linking agent is desired to remain stable until it accesses the target site. As the highly-reactive vinyl group of T-vinyl may suffer from a non-desired attack by ubiquitous nucleophiles, such as water, it is desirable to be masked with a protecting group that may generate the vinyl group in the vicinity of the target site (Fig. 1). The S-pyridinyl protected T-vinyl, which regenerates the vinyl group under acidic conditions, was applied to the intra-strand crosslink formation of the i-motif DNA.16) In this study, we systematically searched for a suitable sulfide protecting group which may regenerate the vinyl group by β-elimination, and applied it to the cross-link with mRNA to investigate its effect on the translation.
The S-Oct derivative was oxidized with magnesium monoperoxyphthalate (MMPP) in carbonate buffer (pH 10), followed by the addition of 0.5 M NaOH, then neutralized with 5% AcOH to produce T-vinyl oligoribonucleic acid (ORN). A solution of a thiol compound in CH3CN was added to a solution of the T-vinyl ORN in MES buffer (1 M NaCl, 0.5 M MES) and the mixture reacted at pH 7 and room temperature for 30 min.
Results and Discussion
Inducible Crosslinking ReactionThe S-Octyl protected T-vinyl was synthesized using thymidine as the starting material and incorporated into the 2′-O-methyl oligoribonucleotide (S-Octyl ORN) according to a previously reported method. The 2′-methoxy ORN was chosen as a platform to avoid cleavage of the target RNA by ribonuclease H (RNase H) in the biological assay. The sequences of ORN1–5 are shown in respective figures. The synthesized S-Octyl ORN was oxidized with MMPP followed by the elimination of the formed sulfoxide derivative by the treatment with 0.5 M NaOH to produce the T-vinyl ORN. Next, the T-vinyl ORN was protected again as the sulfide derivative using thiols; i.e., methanethiol, 4-substituted thiophenol, 2-mercaptopyridine, and 2-mercaptopyrimidine.
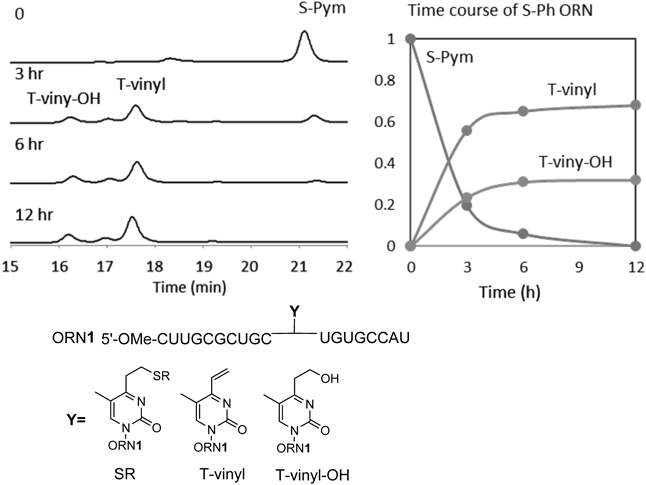
The rate of vinyl formation from the sulfide form was measured by HPLC. Figure 2 illustrates an example of the 2-thiopyrimidinyl protected T-vinyl ORN (S-Pym ORN1). The T-vinyl was readily formed together with the elimination of S-Pym, being associated with the water adduct (T-vinyl-OH). S-Pym ORN1 was converted to T-vinyl (56%) together with T-vinyl-OH (24%) at 3 h, which seemed to be in equilibrium because their ratio did not significantly change. The half-life of the sulfide protected T-vinyl was obtained in a similar manner using the S-alkyl, S-para-substituted phenyl and S-pyridine and S-pyrimidine derivatives, which are subsequently discussed in relation to the cross-linking yield. Tables 1 and 2 summarize the crosslink yields and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF)/MS data of ORNs, respectively.
Table 1. The Cross-Link Yield (%) and the Half-life of the Sulfide Derivative
a)| Run | RS | Cross-link yield [%] | t1/2 [h] |
|---|
| 1 | S-Oct | 3 | 69 |
| 2 | S-Me | 7 | 347 |
| 3 | S-Ph(H) | 50 | 2.5 |
| 4 | S-Ph(Me) | 61 | 5.5 |
| 5 | S-Ph(Br) | 60 | 2.8 |
| 6 | S-Ph(OH) | 61 | 4.1 |
| 7 | S-Ph(NH2) | 60 | 2.9 |
| 8 | S-Ph(NO2) | 35 | 8.5 |
| 9 | S-Ph(COO−) | 50 | 7.9 |
| 10 | S-Pym | 71 | 1.5 |
| 11 | S-Py | 87 | —b) |
a) Conditions for the measurement of the half-life and the cross-linking reaction are described in the footnotes of Figs. 2 and 3. b) The S-pyridine protected T-vinyl could not be isolated due to its instability, therefore, its half-life was not measured.
Table 2. MALDI-TOF/MS Data of the ORN
1–
6 Derivatives
| ORN number | 5′ Modification | Modification of T-vinyl | Calcd. ([MH]−) | Found |
|---|
| ORN1 | — | S-Oct | 6370.19 | 6370.40 |
| ORN1 | — | S-Oct-oxide | 6386.18 | 6389.11 |
| ORN1 | — | T-Vinyl | 6224.07 | 6224.35 |
| ORN1 | — | S-Ph(H) | 6334.09 | 6334.12 |
| ORN1 | — | S-Ph(Me) | 6348.11 | 6348.50 |
| ORN1 | — | S-Ph(Br) | 6412.01 | 6416.18 |
| ORN1 | — | S-Ph(OH) | 6350.09 | 6352.15 |
| ORN1 | — | S-Ph(NH2) | 6349.11 | 6351.64 |
| ORN1 | — | S-Ph(NO2) | 6379.08 | 6374.35 |
| ORN1 | — | S-Ph(COOH) | 6378.09 | 6381.31 |
| ORN1 | — | T-Vinyl-OH | 6242.08 | 6246.59 |
| ORN1 | — | S-Me | 6272.08 | 6272.15 |
| ORN1 | — | S-Pym | 6336.08 | 6337.62 |
| ORN1 | — | S-Py | 6224.07 | 6221.82 |
| ORN3 | FAM | S-Oct | 5533.10 | 5533.29 |
| ORN3 | FAM | T-Vinyl | 5386.99 | 5387.68 |
| ORN3 | FAM | S-Ph | 5497.01 | 5497.61 |
| ORN3 | FAM | S-Pym | 5499.00 | 5498.65 |
| ORN4 | FAM | S-Oct | 5533.10 | 5533.67 |
| ORN4 | FAM | T-Vinyl | 5386.99 | 5586.33 |
| ORN4 | FAM | S-Ph | 5497.01 | 5497.49 |
| ORN4 | FAM | S-Pym | 5499.00 | 5498.24 |
| ORN4a | — | S-Pym | 4961.88 | 4962.84 |
| ORN5 | — | S-Oct | 3696.73 | 3696.69 |
| ORN5 | — | T-Vinyl | 3550.62 | 3550.26 |
| ORN5 | — | S-Pym | 3662.63 | 3664.96 |
| ORN6 | — | S-Oct | 3672.72 | 3672.65 |
| ORN6 | — | T-Vinyl | 3526.61 | 3526.48 |
| ORN6 | — | S-Pym | 3638.62 | 3638.58 |
ORN1: 5′ OMe-CUUGCGCUGCXUGUGCCAU, X = T-vinyl derivative ORN3: 5′ OMe-CUCUGAUUXACGCCC, X = T-vinyl derivative ORN4: 5′ OMe-CUCUGXUUAACGCCC, X = T-vinyl derivative ORN5: 5′ OMe-GUCUUGAXUUC, X = T-vinyl derivative ORN6: 5′OMe-UCCTTGUXGUC, X = T-vinyl derivative
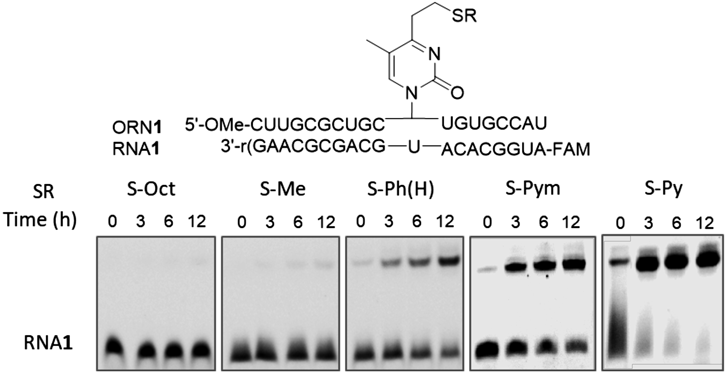
The crosslinking reaction between the ORN1 and RNA1 with U at the complementary site was evaluated in the buffer at pH 7 and 37°C. After 14 h, the reaction was stopped by the addition of a loading buffer containing formaldehyde and ethylenediamine (EDTA), then the mixture was analyzed by denaturing polyacrylamide gel electrophoresis (Fig. 3). The fast mobility and slow mobility bands, RNA1 and the cross-linked adduct, respectively, were quantified and the cross-linking yields were obtained. Table 1 summarizes the crosslink yields between ORN1 and RNA2 together with the half-life of the cross-linking ORN.
The S-Octyl ORN and S-Me ORN were quite stable with the half-lives of 3 d and 14 d, respectively, thus they did not produce any cross-linked product. The thiophenyl derivative (S-Ph(H)) showed a moderate stability (t1/2 = 2.5 h) and formed a cross-link in a moderate yield. Interestingly, both an electron-donating and withdrawing group at the 4-position of the thiophenyl group lengthened the half-life, with representative examples of the methyl group and nitro group (entries 4 and 8). Although it is difficult to rationalize the effect of 4-substituent of the thiophenyl group, it may be speculated based on the protonation of the sulfur atom, which leads β-elimination to produce the vinyl group; the positive charge of the protonated sulfur atom of S-Ph(Me) is partially neutralized by the electro-donating nature of the 4-methyl group, on the other hand, the protonation ability of the sulfur atom of S-Ph(NO2) is weakened due to the electron withdrawing effect of the 4-nitro group. These opposite effects are reflected in the cross-link yield; enhancement by S-Ph(Me) vs a decrease by S-Ph(NO2).
We next investigated sulfide derivatives having 2-thiopyrimidine (S-Pym) or 2-thiopyridine (S-Py). The S-Py ORN regenerated the T-vinyl during purification, therefore, it was subjected to a cross-linking reaction with RNA without isolation and purification. S-Pym ORN showed a lower stability with t1/2 = 2.5 h, nevertheless, it rapidly formed a crosslink in a higher yield. The relatively unstable S-Py showed the fastest reaction and gave the cross-linked product in 85% yield. There is a weak correlation between the cross-linking yield and the half-life of the sulfide derivative; S-Py with a shorter half-life produced the crosslink in the highest yield, while S-Ph(NO2) with the long half-life did in the lowest yield. S-Py ORN was applied to the intrastrand cross-link to stabilize thei-motif. Regarding the balance between the stability and cross-linking reactivity, S-Pym exhibited both a good stability and high reactivity, thus it was used to investigate the effect of the cross-link of mRNA on the translation.
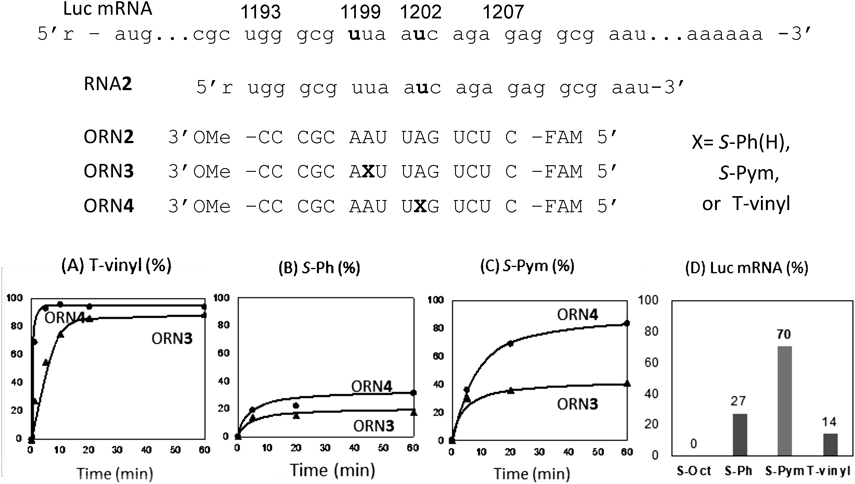
Cross-Linking Reaction with mRNA and Its Effect on the TranslationWe next tested the cross-linking reaction with luciferase (Luc) mRNA to investigate the effect of the cross-link on the translation. This time, the cross-linking ORN labeled with FAM at the 5′ end was used to quantitatively analyze the cross-link yield to mRNA. Prior to the experiment with luc mRNA, the cross-linking efficiency was first checked using the short 27mer RNA2, which corresponds to the region between 1193 and 1207 of the luciferase mRNA. The ORN2 is a control without a cross-linking agent, S-Pym or T-vinyl are placed at X in ORN3 for 1199U and ORN4 for 1202U with 15 nucleotide length. T-vinyl ORN (3 and 4) showed the most efficient reaction to both targets, in particular, the reaction of ORN4 to 1202U was the fastest (Fig. 4A). The S-Ph ORN only slightly formed the adducts with either targets. In the case of the S-Pym ORN4, although the reaction rate was slower than that of T-vinyl ORN4, the cross-linked product gradually increased and reached an 80% yield after 60 min. The difference in the cross-linking efficiency at the target site may be due to the sequence dependency of the T-vinyl derivative, because the non-reactive S-Octyl ORN3 and S-Octyl ORN4 showed similar melting temperatures with the RNA2 (55.8 and 55.9°C, respectively). These ORN4 were next tested for cross-linking with Luc mRNA. The cross-link reaction was done using 3.8 µM ORN4 for 1.1 µM Luc mRNA in MES buffer at 37°C for 3 h under which the conditions of more 90% Luc mRNA was confirmed to form the duplex with ORN4. Unexpectedly, only a low yield was obtained with the T-vinyl ORN4, whereas the S-Pym ORN4 formed the cross-link in a similarly yield with the short RNA2 (Fig. 4D). It is probable that the access of T-vinyl ORN4 to Luc mRNA was delayed because of the sterically large mRNA and thereby undesired reactions such as water attack to the active vinyl group were caused. Thus, the ORN4 was next used for the investigation of the effect on the translation of luc mRNA.
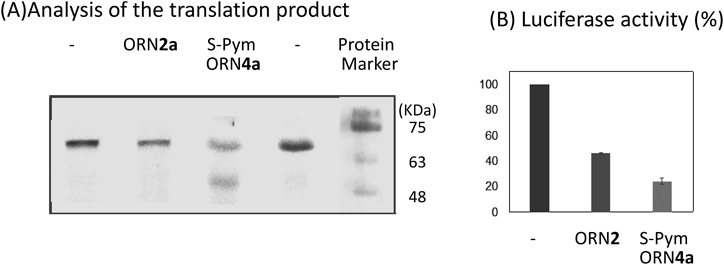
The Luc mRNA was incubated for 6 h in MES buffer at 37°C and pH 7 in the presence of S-Pym ORN4a (the same sequence with ORN4 except for the 5′ FAM label) or non-reactive ORN2a (the same sequence with ORN2 except for the 5′ FAM label) as the control. Translation was carried out at 25°C for 2 h using the wheat germ extract, and the luciferase activity was evaluated by measuring the luminescence. In addition, the translation products were analyzed by 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) electrophoresis. ORN2 reduced both the production of the luciferase and its activity to about 50% due to the steric block by ORN2. In contrast, S-Pym ORN4 reduced the luciferase to less than 50% associated with the truncated proteins (Figs. 5A, B). The reduced luciferase activity might occur from the non-reacted Luc mRNA. According to the estimated molecular weight of about 45 kDa, the fast moving bands suggested that the truncation occurred at the cross-linked site (Fig. 5A). It was also reported that the translation of the cross-linked Luc mRNA is terminated at the codon before the crosslink site, however, the precise truncated position was not clear.17)
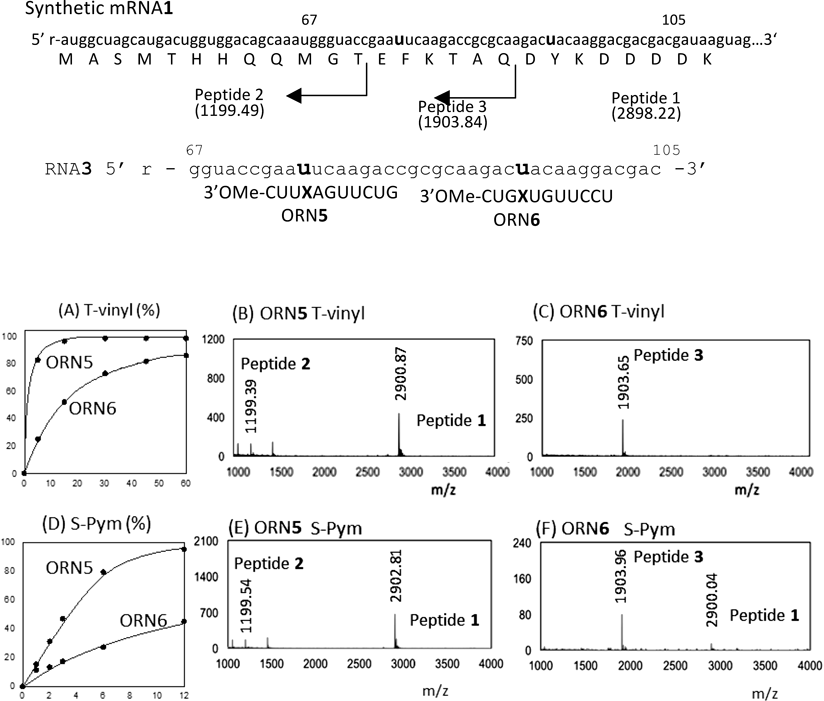
To precisely determine the truncation site in the translation from the cross-linked mRNA, we next performed the translation using the synthetic mRNA. The synthetic mRNA has a 131 nucleotide length, which codes 26 mer Peptide 1 with 2898.22 Da containing the T7 tag and the Flag tag. The cross-link reaction was first checked using 39 mer RNA which corresponds to the region between 67 and 105 of the synthetic mRNA (Fig. 6). ORN5 and ORN6 target 76U and 94U, respectively. The reactivity tendency of the T-vinyl and S-Pym ORN is retained in that T-vinyl reacts faster than S-Pym (Figs. 6 A, B). In this sequence, ORN5 produced higher cross-link yields with both the T-vinyl and S-Pym derivatives. Next, the synthetic mRNA was subject to the cross-link formation with ORN for 12 h at 37°C and pH 7. The reacted mRNA was translated using the PURE system (PUREflex® 2.0) for 2 h at 37°C. The produced peptides were collected using Anti-T7-tag agarose beads, and were analyzed based on MALDI-TOF/MS measurements. Non-cross linking ORN containing adenosine at X in ORN5 and ORN6 did not inhibit the translation, and the full-length Peptide 1 was produced. On the other hand, T-vinyl ORN5 induced the truncated Peptide 2, which indicated that the translation stopped at the 72-adenosine. This fact implies that the translation reaction stopped at the end of the cross-linked ORN5. Similarly, T-vinyl ORN6 induced the truncated Peptide 3, clearly indicating that the translation stopped at 90-guanosine, the end of the cross-linked ORN6. Interestingly, although the crosslink rate of T-vinyl ORN6 was slower than that of T-vinyl ORN5 for RNA3, peptide 3 was produced as a major product by T-vinyl ORN6 (Fig. 6, B vs C). ORN5 and ORN6 with the S-Pym derivative also showed a similar tendency in the production of the truncated peptides 2 and 3 depending on the cross-linked sites, which were also an indication of the translation stop at the duplex formed by the cross-linked ORN. These results have clearly indicated that the duplex formed between mRNA and ORN is sufficiently stabilized by the cross-link to stop the translation at the end of the formed duplex.
Conclusion
The 4-vinylpyrimidin-2-one nucleoside (T-vinyl) forms a cross-link with the RNA containing uracil at the complementary site at a high reaction rate. To obtain the stable T-vinyl derivative so that its reactivity is protected until it access to the target site, several derivatives were investigated, and the 2-thiopyridinyl- and 2-thiopyrimidinyl T-vinyl derivatives were determined to be good candidates. The 2-thiopyridinyl derivative was applied to cross-link the i-motif.16) The 2-thiopyrimidinyl T-vinyl derivative was found to more efficiently cross-link with mRNA albeit having a better stability than the 2-thiopyridinyl T-vinyl derivative. The investigation using the Luc mRNA, the synthetic mRNA and non-cellular translation system revealed that the translation is terminated at the end of the cross-linked duplex between the mRNA and the ORN. Thus, the 2-thiopyrimidinyl T-vinyl derivative has successfully demonstrated both a good stability and high efficiency for the cross-linking reaction, and expanded its applicability in biological applications.
Experimental
The Synthesis of the 2′-OMe ORN Incorporating the S-Octyl Protected T-Vinyl: S-Oct ORN3The 3′-O-[2-cyanoethoxy (diisopropylamino) phosphino]- 5′-O-(4,4′-dimethoxytrityl)- 4-(2-octylthioethyl) derivative of thymidine was prepared according to a previously reported method, which was applied to the automated DNA synthesizer for incorporation into 2′-OMe ORN. The synthesis was performed using the standard protocol of the amidite chemistry on a 1 µmol scale, and the synthesized oligonucleotide was cleaved from the CPG resin using K2CO3 (45 mM) and octanethiol (1 mM) in dry methanol (1 mL) for 6 h at room temperature. The 5′-O-DMTr protected oligonucleotide was purified by HPLC (Column: SHISEIDO CAPCELL PAK C18 MG, 4.6 × 250 mm; Solvent A: 0.1 M triethylammonium acetate (TEAA) Buffer, Solvent B: CH3CN, B: 20% to 30% /20 min, 30% to 100%/25 min, linear gradient; Column oven: 50°C; Flow rate: 1.0 mL/min; UV: 254 nm) and the collected fraction was freeze-dried. The isolated product was treated with 10% aqueous AcOH to deprotect the DMTr group, then purified again by HPLC (Column: SHISEIDO CAPCELL PAK C18 MG, 4.6 × 250 mm; Solvent A: 0.1 M TEAA Buffer, Solvent B: CH3CN, B: 20% to 50%/20 min, 50% to 100%/25 min, linear gradient; Column oven: 50°C; Flow rate: 1.0 mL/min; UV: 254 nm; Column: SHISEIDO CAPCELL PAK C18 MG, 4.6 × 250 mm; Solvent A: 0.1 M TEAA Buffer, Solvent B: CH3CN, B: 20% to 50% /20 min, 50% to 100%/25 min, linear gradient; Column oven: 50°C; Flow rate: 1.0 mL/min; UV: 254 nm). The synthesized ORN was subject to structure determination by MALDI/TOF MS and the quantification by UV-Vis measurement at 260 nm.
The Synthesis of the Sulfide Protected T-Vinyl ORNA solution of magnesium monoperoxyphtalate hexahydrate (MMPP) in a carbonate buffer (0.5 mM, 0.3 µL, 150 pmol) was added to a solution of S-Ocy ORN in (152 µM, 0.33 µL, 50 pmol) in a carbonate buffer (125 mM, 0.2 µL, pH 10), and the mixture was stored for 30 min followed by the addition of an aqueous NaOH solution (4 M, 0.21 µL). After 1 min, the mixture was neutralized with an aqueous 25% AcOH solution to give the corresponding T-vinyl ORN, which was used for the next reaction without purification.
A solution of a thiol compound in CH3CN (1 mM, 20 µL, 20 nmol) was added to a solution of T-vinyl ORN (26 µM, 75.9 µL, 2 nmol) in MES buffer (1 M NaCl, 0.5 M MES, 12.5 µL, pH 7), and the mixture was diluted with MilliQ to adjust the following final concentrations; T-vinyl ORN (16 µM), MES buffer (50 mM), NaCl (100 mM), thiol compound (160 µM). After 30 min, the sulfide protected T-vinyl ORN was isolated by HPLC (Column: SHISEIDO CAPCELL PAK C18 MG, 4.6 × 250 mm; Solvent A: 0.1 M TEAA Buffer, Solvent B: CH3CN, B: 10% to 15%/20 min, 15% to 100%/25 min, linear gradient; Column oven: 50°C; Flow rate: 1.0 mL/min; UV: 254 nm). The synthesized ORN was subject to structure determination by MALDI/TOF MS and the quantification by UV-Vis measurement at 260 nm.
The Cross-Linking Reaction with RNAA reaction mixture containing T-vinyl ORN or the sulfide protected T-vinyl ORN (5 µM), RNA (1 µM), and MES buffer (100 mM NaCl, 50 mM MES, pH 7) was reacted at 37°C, and a portion the reaction mixture (1.9 µL) was added to a loading buffer (1.9 µL) at a specific time interval and subjected to the analysis by gel electrophoresis using 15% denatured polyacrylamide gel (2 h a 250 V). The 15% polyacrylamide gel was prepared using urea (7 M and 1 × TBE buffer). Loading buffer: 95% formamide, EDTA (20 mM), BPB (0.05%), XCF (0.05%), TBE buffer: Tris–HCl (89 mM), boric acid (89 mM), EDTA (2 mM), pH 8.0.
The Duplex Formation, the Cross-Linking Reaction with Luciferase mRNA and the Following TranslationThe mixture containing luc mRNA (1.1 µM), RNase OUT (4 u/µL), S-Oct ORN (0.38–7.8 µM), MES buffer (50 mM) and NaCl (100 mM) was heated at 60°C for 5 min, then kept at 37°C for 1.5 h. The formation of the duplex was analyzed using a 2% non-denatured agarose gel.
A similar procedure was applied to the cross-linking and following translation. A solution was prepared in 2 µL containing Luc mRNA (0.5 µM), RNase OUT (2 u/µL), T-vinyl, S-Pym ORN (5 µM), MES buffer (50 mM) and NaCl (100 mM), and heated at 60°C for 5 min, then kept at 37°C for 6 h. To this solution were added an aqueous AcONa (1 M, 0.5 µL, 0.5 µmol), amino acid mixture (1 mM, 0.4 µL, 0.4 nmol), Fluoro Tect™ GreenLys tRNA (0.3 µL), Wheat Germ Extract (2.5 µL) at 0 C, then the mixture was incubated at 25°C for 2h. The final concentrations in the translation mixture were Luc mRNA (0.18 µM), ORN (1.8 µM), AcONa (88 mM) and amino acid mixture (70 µM). The reaction was stopped by the addition of the loading buffer (5.7 µL) and heating at 70°C for 3 min, then analyzed using a 20% SDS-polyacrylamide gel (20 mA, 2 h and 20 min) and WIDE-VIEW™. Prestained Protein Size Marker III (Wako) was used as a marker. The gel image was visualized by LAS-4000 (λEx = 494 nm, λEm = 518 nm). Running gel: 20% polyacrylamide, 0.1% SDS, Tris–HCl (0.375 M), pH 8.8. Stacking gel: 4.75% polyacrylamide, 0.1% SDS, Tris–HCl (0.125 M), pH 6.8. Loading buffer: Tris–HCl (0.1 M), 4% SDS, 2-mercaptoethanol (1.7 M), glycerol (2.7 M), 0.05% BPB.
Luciferase AssayReagent (Promega, 15 µL) was added to the translation mixture, and the luciferase activity was measured by a GloMax® 20/20 Luminometer.
The Cross-Linking with the Synthetic mRNA and the Translation of the Cross-Linked mRNAThe all-sequence of the synthetic mRNA is shown below, in which the underlined aug and uag represent the start and stop codon, respectively. The double underlined u represent the target uracil base for ORN5 and ORN6.
5′-ggg ucu aga guu uaa cuu uaa gaa gga gau aua cau aug gcu agc aug acu ggu gga cag caa aug ggu acc gaa uuc aag acc gcg caa gac uac aag gac gac gac gau aag uag uga aua acu aau cc-3′
ORN5: 3′OMe-CUUXAGUUCUG, X = T-vinyl or S-pym.
ORN6: 3′OMe-CUGXUGUUCCU, X = T-vinyl or S-pym.
Peptide 1: MASMTGGQQMGTEFKTAQDYKDDDDK (calculated MS (m/z) for [M + H]: 2898.2231).
Peptide 2: MASMTGGQQMGT (calculate MS (m/z) for [M + H]: 1199.4859).
Peptide 3: MASMTGGQQMGTEFKTAQ (calculate MS (m/z) for [M + H]: 1903.8352).
A solution containing the synthetic mRNA (5 µM) and ORN (20 µM) in MES buffer (50 mM) containing 100 mM NaCl was reacted for 12 h at 37°C and pH 7. After dilution with Milli-Q, the reacted mRNA was collected by centrifugal filtration (Amicon Ultra-0.5 mL 10K; Merck). The collected mRNA (2 µg/7 µL) was dissolved in Solution I, Solution II and Solution III (2 µL) provided in an in vitro translation kit (PUREflex® 2.0), and the translation was performed for 2 h at 37°C. The NP-40 Lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 0.05% NP-40, pH 7.5, 1330 µL) was added to the translation solution (20 µL), followed by the addition of Anti-T7 tag agarose pAB (MBL, 15 µL). The solution was shaken for 12 h at 0 C, centrifuged at 2500 × g and 4°C for 3 min, then the supernatant was removed. This procedure was repeated twice. The collected beads were suspended in ice-cold 50% aqueous CH3CN-0.1% formic acid (75 mL), centrifuged at 2500 × g and 4°C for 3 min, the supernatant was collected, then this procedure was repeated twice. The eluted fractions (150 µL) were diluted with 50 mM formic acid buffer (1050 µL), lyophilized and desalted with ZipTip. The isolated peptides were analyzed based on MALDI-TOF/MS measurements. The same procedure was done with the non-cross linking ORN containing adenosine at X in ORN5 and ORN6.
Acknowledgments
We are grateful for the support provided by a Grant-in-Aid for Scientific Research (B) (No. 18H02558) from the Japan Society for the Promotion of Science (JSPS).
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Shen X., Corey D. R., Nucleic Acids Res., 46, 1584–1600 (2018).
- 2) Goyal N., Narayanaswami P., Muscle Nerve, 57, 356–370 (2018).
- 3) Bennett C. F., Annu. Rev. Med., 70, 307–321 (2019).
- 4) Yamayoshi A., Kato K., Iwase R., Yamaoka T., Wake N., Murakami A., Nucleic Acids Res., 3 (Suppl.), 75–76 (2003).
- 5) Higuchi M., Yamayoshi A., Yamaguchi T., Iwase R., Yamaoka T., Kobori A., Murakami A., Nucleosides Nucleotides Nucleic Acids, 26, 277–290 (2007).
- 6) Johansson H. E., Helsham G. J., Sproat B. S., Hentze M. W., Nucleic Acids Res., 22, 4591–4598 (1994).
- 7) Sakamoto T., Fujimoto K., “Photo-Cross-Linking Reaction in Nucleic Acids: Chemistry and Applications”, Chap. 7, “Modified Nucleic Acids” ed. by Nakatani K, Tor Y., Springer, 2016, pp. 145–158.
- 8) Shigeno A., Sakamoto T., Yoshimura Y., Fujimoto K., Org. Biomol. Chem., 10, 7820–7825 (2012).
- 9) Fujimoto K., Konishi-Hiratsuka K., Sakamoto T., Yoshimura Y., ChemBioChem, 11, 1661–1664 (2010).
- 10) Op de Beeck M., Madder A., J. Am. Chem. Soc., 134, 10737–10740 (2012).
- 11) Liu Y., Rokita S. E., Biochemistry, 51, 1020–1027 (2012).
- 12) Nagatsugi F., Kawasaki T., Usui D., Maeda M., Sasaki S., J. Am. Chem. Soc., 121, 6753–6754 (1999).
- 13) Kawasaki T., Nagatsugi F., Ali M. M., Maeda M., Sugiyama K., Hori K., Sasaki S., J. Org. Chem., 70, 14–23 (2005).
- 14) Ali M. M., Oishi M., Nagatsugi F., Mori K., Nagasaki Y., Kataoka K., Sasaki S., Angew. Chem. Int. Ed., 45, 3136–3140 (2006).
- 15) Nishimoto A., Jitsuzaki D., Onizuka K., Taniguchi Y., Nagatsugi F., Sasaki S., Nucleic Acids Res., 41, 6774–6781 (2013).
- 16) Kikuta K., Piao H., Brazier J., Taniguchi Y., Onizuka K., Nagatsugi F., Sasaki S., Bioorg. Med. Chem. Lett., 25, 3307–3310 (2015).
- 17) Hagihara S., Kusano S., Lin W. C., Chao X. G., Hori T., Imoto S., Nagatsugi F., Bioorg. Med. Chem. Lett., 22, 3870–3872 (2012).