Abstract
Fibrillated aggregation of amyloid β (Aβ) peptides is a potential factor causing toxic amyloid deposition in neurodegenerative diseases. A toxic fibril formation of Aβ is known to be enhanced on the ganglioside-rich lipid membrane containing some amounts of cholesterol and sphingomyelin. This ganglioside-rich membrane is supposed to provide a hydrophobic environment that promotes the formation of Aβ fibrils. Molecular dynamics simulations were carried out to investigate the structure of Aβ complex in the hydrophobic solution composed of dioxane and water molecules. The Aβ conformation was contrasted to that in the aqueous condition by executing multiple computational trials with the calculation models containing one, four, or six Aβ peptides. The conformation was also compared between the calculations with the 42-mer (Aβ42) and 40-mer (Aβ40) peptides. The simulations for Aβ42 demonstrated that Aβ peptides had a tendency to stretch out in the hydrophobic environment. In contrast, Aβ peptides were closely packed in the aqueous solution, and the motions of Aβ peptides were suppressed significantly. The N-terminal polar domains of Aβ peptides tended to be positioned at the inside of the Aβ complex in the hydrophobic environment, which supported the C-terminal domains in expanding outward for inter-molecular interaction. Since Aβ peptides were not tightly packed in the hydrophobic environment, the total surface area of the Aβ complex in the hydrophobic solution was larger than that in the aqueous one. The simulation for Aβ40 peptides also showed a difference between the hydrophobic and aqueous solutions. The difference was compatible with the results of Aβ42 in the structure of the Aβ complex, while the C-terminal outward expansion was not so distinct as Aβ42 peptides.
Introduction
Amyloid β (Aβ) peptide is known as a major constituent of senile plaques in the brains of patients of Alzheimer’s disease (AD).1) The Aβ polymerization into fibrils has been broadly investigated because it is a critical step in the pathological process of the neurodegenerative disorders like AD.2) It has been suggested that, in the case of the absence of membrane, Aβ peptides develop into fibrils through a nucleation polymerization pathway that is characterized by an initial monomer-dominant phase, followed by a conformational shift to a β-sheet and an assembly of monomers into oligomers.3) In this pathway, oligomers are nuclei for the fibril aggregation, and the nuclei elongate through monomer addition to the growing polymers according to the so-called nucleation and growth process.4) In an experiment,5) the Aβ nucleation was observed at high Aβ concentrations in the range of 17.5–100 µM, while the physiological concentration of Aβ in the human brain is considered to be in the sub-nM level. Namely, the Aβ concentration is lower than the value set in the in vitro experiment without membrane by several orders. This means that the Aβ fibrillation will follow an alternative mechanism in vivo. Several studies have suggested a template-assisted mechanism to lower the energy barrier for the Aβ nucleation.6) In the mechanism, the cell membrane containing GM1 ganglioside provides a unique local environment and works as a catalyst for fibril formation.7) Indeed, the presence of GM1-containing neuronal cell membranes facilitated the polymerization of Aβ42 and Aβ40 at nanomolar concentrations.8)
In the outer leaflet of a cell membrane, GM1 gangliosides compose clusters with including sphingomyelin (SM) and cholesterol (Chol) at high concentrations.9) This cluster is one of the major components of a particular kind of microdomain that has been identified as a detergent-insoluble glycosphingolipid-rich component in the membrane of brain cells.10) GM1 ganglioside is abundant in the particular microdomain. The enhancement of Aβ binding to the cell membrane and the acceleration of the subsequent fibril formation were observed on the GM1 ganglioside containing microdomain.11) The concentrations of GM1 ganglioside and Chol are critical factors in the Aβ aggregation on the GM1 ganglioside-containing membrane and have strong influences on the fibrillation rate.12)
The amyloid fibrils formed on the cell membrane are cytotoxic, while those formed in aqueous solution are not.13) Hence, the presence of GM1 ganglioside will be significantly important for the aggregation of toxic Aβ fibrils. Based on the assumption that low-polarity environment provided by GM1 clusters promotes the formation of toxic Aβ fibrils, Fukunaga et al. investigated the structures of Aβ aggregates in solutions with several different polarities by mixing 1,4-dioxane into water.14) They found that a mixture of 80% dioxane and 20% water gave the polarity similar to the membrane surface, and the morphology, secondary structure, and cytotoxicity of the Aβ fibrils formed on this mixed solution were close to those of the toxic Aβ one.
To grasp the growth process of the toxic Aβ fibrillation, simulation analysis should be performed on the structural transfer from the Aβ gathering into the Aβ nucleus that will develop into the toxic amyloid fibrils. In this work, we carried out long-time molecular dynamics (MD) simulations using the calculation models including one, four, or six Aβ peptides in the hydrophobic environment composed of the mixture of 1,4-dioxane and water. For reference, MD simulations were also carried out in the aqueous condition. The structures of the Aβ aggregates were monitored through the simulations. Clarifying the molecular mechanism of the embryonic stage of Aβ fibril will be significantly important for designing an inhibitor that blocks the Aβ fibrillation or the deposition of Aβ plaque.
Experimental
Construction of Calculation ModelsOne, four, and six Aβ1–42 peptides were separately placed in a rectangular box filled with 1,4-dioxane and water molecules in a ratio of 4 : 1 as models for the low-polarity solution. For comparison, one, four, and six Aβ peptides were also separately placed in a box solvated with water. The initial positions of Aβs were manually set to make the closest atom distance between any two Aβ molecules more than 5.0 Å. Sodium and chloride ions were also contained in the model to make the ion concentration equal to 150 mM. The final model size was ca. 92 × 92 × 93 Å, and the total number of atoms was about 80000 for every Aβ model. The initial atom coordinates of Aβ1–42 were extracted from a protein data bank (PDB)-deposited structure: 1Z0Q, which had been determined by solution NMR spectroscopy.15) The structure 1Z0Q contains 30 conformations, and the 11th one was selected for the initial coordinates of Aβ for a reason described elsewhere.16)
We also built calculation models with Aβ40. Six Aβ1–40 peptides were placed both in a box filled with a mixture of 80% 1,4-dioxane and 20% water and in a box solvated with water. The initial atom coordinates of Aβ40 were extracted from the structure with a PDB code: 2LFM.17) This structure was determined by solution NMR spectroscopy without any detergent. The structure, 2LFM, contains 20 conformations, and the first one was selected.18)
Computational Condition for MD SimulationMinimization, heating, and pre-equilibration were executed using the sander module of AMBER16.19) The production run of MD simulation was carried out using the pmemd module. Energy minimization was achieved in two steps. The first step was subject to relaxation of the Aβ molecules only. The second step was for relaxation of the whole system. In each step, energy minimization was done for 10000 cycles. The minimization method was switched from the steepest descent method to the conjugate gradient method after 3000 cycles. The model system was heated to 310 K for 0.1 ns under the NVT-ensemble condition, and subsequent 0.4 ns pre-equilibrating calculation was executed under the NPT-ensemble condition. Then production runs of MD simulation were carried out for 1 µs. Only for the six Aβs models, the simulation was extended to 5 µs. For Aβ40, simulations were carried out only with the six peptide model and the simulation time was 5 µs. The cutoff distance for the electrostatic and van der Waals energy terms was set to 12.0 Å, and the periodic boundary condition was applied. The particle mesh Ewald method was employed to calculate the long-distance electrostatic force. The expansion and shrinkage of every covalent bond connecting to a hydrogen atom were constrained. The integration time step was 2 fs.
The ff14SB force field20) was applied to the protein molecules, while the general AMBER force field 2 (GAFF2)21) was to 1,4-dioxane. The atom charges of 1,4-dioxane were determined by the approach making use of the quantum chemical calculation.22,23) The stable structure of 1,4-dioxane was initially determined through geometry optimization at the B3LYP/6-31G(d,p) level, and the electrostatic potential was subsequently calculated at the B3LYP/cc-pVTZ level. The atom changes were calculated from the electrostatic potential.
Analysis of Simulation ResultsThe ptraj module of AmberTools1620) was utilized to obtain snapshot structures from the simulation trajectory. The snapshot structures were depicted by PyMOL.24) For a better understanding of simulation structures, the positions of some Aβ molecules were transferred along x, y, or z-direction by the side length of the periodic boundary box.
Cluster analysis on the conformational diversity of Aβ42 and Aβ40 was performed in a manner similar to our previous work.25) The coordinates of main-chain atoms were extracted every 0.1 µs from the trajectory of the last 1 µs for the six Aβs models. The average structure of the extracted structures was obtained. Then each extracted structure was fitted to the average one to calculate the root-mean-square deviation (RMSD). Based on the RMSD values, the structures were classified by performing cluster analysis with the nearest neighboring method. Finally, all of the structures were connected as a tree called a dendrogram, in which the x-axis is the labels for the snapshot structures and the y-axis is the distance for the least dissimilarity among the individual structures. Based on the dendrogram, the snapshot structures were separated into several groups with setting the criteria distance to 15 Å.
The secondary structure of Aβ peptides was calculated using the DSSP program.26) To monitor the change in secondary structure during simulation, the coordinates of main-chain atoms were extracted every 0.1 µs from the 5 µs trajectories of the six Aβs models both for Aβ42 and Aβ40.
Results
Conformations of Aβ42 Peptides in the Low-Polarity SolutionFigure 1 shows the final structures of the 1 µs MD simulation for one, four, and six Aβ42 peptides in the mixed solution of dioxane and water. Simulation structures of Aβ42 peptides in the aqueous solution are also shown for comparison. As for the single Aβ model, the peptide was unfolded in the mixed solution (Fig. 1a). The secondary structure of the peptide was mainly coil except for one small helix. The polar N-terminal domain of Aβ was positioned inside, and the apolar C-terminal domain was located outside. Aβ peptide was slightly unfolded in the aqueous solution, and the β-sheet structure was formed at the middle of the peptide (Fig. 1a′). No clear difference was observed between the N-terminal and C-terminal domains.
The final structures of the 1 µs simulations for four Aβ peptides also suggest that the folding of Aβ is significantly loosened in the mixed solution (Fig. 1b). All the Aβs were loosely assembled to make a single bunch of peptides. The apolar C-terminal domains of the peptides were likely to be positioned outside of the bunch. In contrast, four Aβs tightly gathered in the aqueous solution (Fig. 1b′). The C-terminal hydrophobic regions were positioned at the middle of the peptide complex. All the four Aβs had a short helix, and those helices were located outside of the peptide complex.
The difference in peptide conformation between the mixed and aqueous solutions was more distinctly observed in the six Aβs models (Fig. 1c). In the mixed solution, the folding was loose for every Aβ, while six Aβs were assembled to be a single complex. The C-terminal apolar domains were positioned outside, and the N-terminal polar domains were inside. The secondary structure of the C-terminal domain was the coil, and then domains were likely to stretch outward. In contrast, the secondary structure of the N-terminal domain was the helix, and the domains were folded compactly. In the aqueous solution, six Aβs gathered and were tightly packed (Fig. 1c′). The helix regions were mostly located outside, and three long helices were observed in the outer area of the peptide assembly.
Extended Simulation of Six Aβ42 PeptidesMD simulations for six Aβs models were extended to 5 µs both for the mixed and aqueous solutions. The final snapshot structures are illustrated with different colors among the Aβs and also a gradation of color from N- to C-terminus for every Aβ (Figs. 2a and 2a′). The structure of Aβ complex in the mixed solution was almost consistent with that of the 1 µs simulation in Fig. 1c. The C-terminal domains of Aβ peptides were positioned outside and spread in the mixed solution. In contrast, the N-terminal polar domains were located on the inside of the Aβ assembly. In the aqueous solution, Aβs were tightly packed, and most of the helix regions tended to gather and kept stable interactions among them.
The structure change of the Aβ assembly over time is shown in Figs. S1 and S2 for the mixed solution and Figs. S3 and S4 for the aqueous one. The six Aβs in the mixed solution were not in a completely single cluster at 0.5 µs (Fig. S1a). All the Aβs gathered at 1.0 µs (Fig. S1b), and the structure of the Aβs complex was not largely changed until 2.0 µs (Figs. S1c and S1d). The C-terminal domains were positioned outside as seen in Figs. S2b–S2d. A few Aβs were converted into an expanded form after 2.5 µs (Fig. S1e), and most of the expanded regions were C-terminal domains (Fig. S2e). The C-terminal domains were further expanded after 3.0 µs (Figs. S1f–S1h), and it was clearly observed in Fig. S2h that the C-terminal domains stretched while the N-terminal domains were compact inside. The expanded C-terminal domains sometimes changed their conformations to the β-bridge after 4.0 µs (Figs. S1i–S1j). Figures S2h–S2j demonstrated that the C-terminal apolar domains were located outside, and the N-terminal polar domains were inside. In contrast to the mixed solution, the structure of the Aβs complex was not drastically altered for 5.0 µs in the aqueous solution. All the Aβs gathered to be one cluster at 0.5 µs (Fig. S3a). The complex structure and the relative positions of the Aβ molecules were hardly altered for 5.0 µs (Figs. S3b–S3j). The C-terminal hydrophobic domains were not always located outside, while the N-terminal polar domains were frequently exposed to solvent (Figs. S4a–S4j).
Since the MD simulations were performed under the periodic boundary condition, the expanded C-terminal domain reaches the boundary edge and possibly interacts with the Aβ peptides of the adjacent calculation cell. Figure 2(b) is an illustration of two Aβ complex in two adjacent calculation cells of the simulation in the mixed solution, in which the Aβ assembly of the adjacent cell was generated by copying the Aβs of the original cell and shifting their coordinates by the length of one unit cell. An expanded C-terminal domain was found to form a parallel β-sheet with the C-terminal domain of another Aβ in the adjacent cell. This β-sheet formation was so stable that it was maintained for the last 2.0 µs of the simulation.
Simulation of Six Aβ40 PeptidesMD simulations were carried out for the models of six Aβ40 peptides in the mixed and aqueous solutions. The final snapshot structures of the 5 µs simulations are shown in Fig. 3. Aβs in the mixed solution were sparsely distributed compared to those in the aqueous solution. The C-terminal domains tended to be in the helix conformation instead of the coil, which is a marked difference from the simulations of Aβ42 peptides. The C-terminal helices were likely to be located on the outside of the Aβ assembly. The N-terminal polar domains were likely to be positioned inside. In the aqueous solution, six Aβs were closely packed with the C-terminal apolar domains positioned inside. The time course of changes in the structure of the Aβ complex is shown in Figs. S5 and S6 for the mixed solution and Figs. S7 and S8 for the aqueous one. Six Aβ40 molecules gathered to be one cluster at 1.0 µs in the mixed solution (Fig. S5b). The relative positions of Aβs were scarcely changed after 1.0 µs (Figs. S5d–S5j). Although Aβs were not closely packed, the C-terminal domains were likely to be helix instead of the coil (Figs. S6b–S6j). Hence, the chance for the C-terminal domain of Aβ40 to interact with other molecules was low compared to the case of Aβ42. In the aqueous solution, Aβ40 peptides gathered to be a single cluster at 0.5 µs (Fig. S7a), and the relative positions of the Aβs were hardly changed for 5.0 µs (Figs. S7b–S7j). The N-terminal domains tended to be located outside to wrap the C-terminal domains inside.
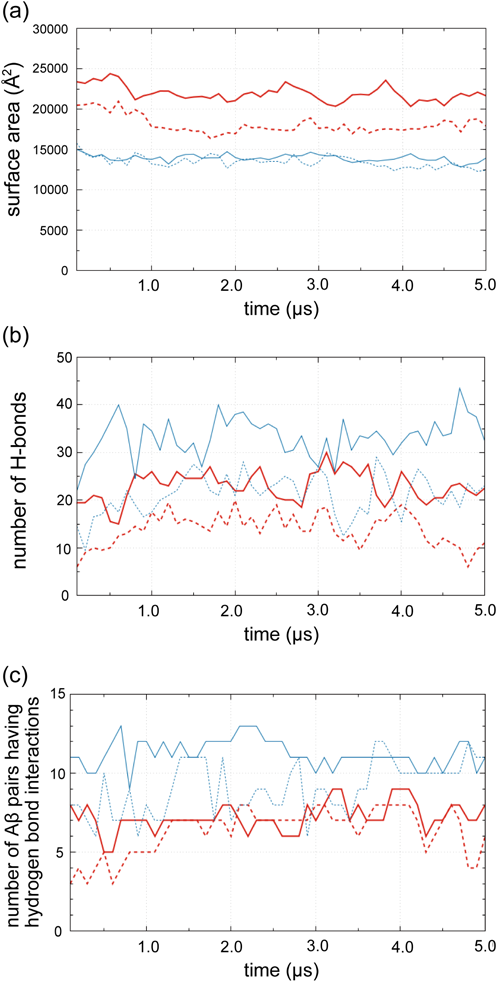
Surface Area of the Aβ Complex and Inter-molecular Hydrogen BondingFigure 4a shows the change in total surface area of the Aβ complex for 5 µs MD simulations with six peptide models in the mixed and aqueous solutions. For both of Aβ42 and Aβ40, the Aβ complex in the mixed solution has larger surface areas than those in the aqueous solution. The difference in surface area between Aβ42 and Aβ40 is slight in the aqueous solution. In contrast, the difference between Aβ42 and Aβ40 is large in the mixed solution. These results mean that the Aβ42 peptides in the mixed solution are most loosely packed among the four models. The loose packing is advantageous for converting the conformations of Aβ peptides. In every model, the surface area tends to decrease over time. Hence, it is expected that the structure of Aβ complex further alters if the simulation time is extended more.
The changes in the total number of inter-molecular hydrogen bonds summed up for all the combinations of every two Aβs are shown in Fig. 4b. For both Aβ42 and Aβ40, the total number of hydrogen bonds in the aqueous solution is larger than that in the mixed solution. This result is compatible with the findings that the Aβ complex in the aqueous solution is more tightly packed compared to that in the mixed solution. The number of inter-molecular hydrogen bonds of every Aβ peptide was also monitored along the simulation (Fig. S9). The numbers ceaselessly vary during the simulation, which implies the possibility that the structure of the Aβ complex is converted if the simulation time is extended much more. The number of Aβ peptide pairs that have hydrogen bond interactions is shown in Fig. 4c. The total number of the Aβs combinations is 15 for six peptides model. About half of the combinations have hydrogen bonds for Aβ42 in the mixed solution, while more than two-thirds of the combinations have hydrogen bonds in the aqueous solution. Similarly, the number of combinations having hydrogen bonding in the aqueous solution is larger than that in the mixed one for Aβ40. This result reflects the situation that the Aβ complex is loosely packed in the mixed solution.
Discussion
Secondary Structure of Aβ PeptidesAβ peptides aggregate into insoluble fibrils, which accumulate to be toxic plaques causing the neurodegeneration.27) According to a solution NMR study, Aβ peptides in the fibrils have the β-sheet conformation and are aligned to be a closely packed form.28) Since the major secondary structure of the isolated Aβ is α-helix,15,29) the conformational conversion of Aβ peptide due to the interaction with other Aβs is expected. Hence, the change in secondary structure was examined for the respective residues of the Aβs through MD simulations (Figures S10–S13).
As seen in Figs. S10 and S11, turn or bend was the major secondary structure of Aβ42 both in the mixed and aqueous solutions. In the mixed solution (Fig. S10), the amount of helix was much larger than that of β-strand or β-bridge. The frequency of β-strand or β-bridge was slightly increased with the progress of MD simulations, and that of β-strand or β-bridge for two Aβs were almost comparable to the helix one [Figs. S10e and S10f]. In the aqueous solution (Fig. S11), a small amount of β-strand or β-bridge constantly appeared, and except for that, no remarkable difference was observed between the mixed and aqueous solutions.
As for Aβ40, helix was the most major secondary structure in the mixed solution, and β-strand or β-bridge was barely observed (Fig. S12). The helix content was quite large. In the aqueous solution, β-strand or β-bridge was observed to some degree, and the helix content was less than that in the mixed solution due to the appearance of β-strand or β-bridge (Fig. S13).
The secondary structure was determined by the DSSP program, which does not take the intermolecular interaction into account. Many intermolecular hydrogen bonds were observed among the Aβ42 peptides in the mixed solution, and most of the residues involving the intermolecular hydrogen bonds formed the turn or bend. The conformation of these residues is expected to be converted into the β-strand with the development into fibrils.
Structural Diversity of Aβ Peptides in SimulationIn the dendrogram obtained by the cluster analysis, the snapshot structures were separated into six major groups and two minor fractions for the last 1 µs of the simulation with six Aβ42 peptides in the mixed solution (Fig. S14a). Every major group almost consisted of the snapshots of a specific one of the six Aβs. This indicated that the conformations of the respective Aβs were no longer largely changed for the last 1 µs, and that the conversion of the Aβ conformation rarely occurred in the Aβ assembly. For the simulation of the aqueous solution, the snapshots of six Aβs were separated into seven groups (Fig. S14b). One Aβ peptide labeled as b was split into two groups, in which the snapshots for 4.2–4.5 µs formed one cluster, and the snapshots for 4.6–5.0 µs formed another. Hence, a noticeable conformational change appeared only for one Aβ for the 1 µs simulation time.
The y-axis of the dendrogram indicates the dissimilarity of the snapshots. The dissimilarities among the group members in the mixed solution are, on the whole, larger than those in the aqueous solution. This means that the motions of Aβ42 peptides are restricted in the aqueous environment compared to the mixed solution.
In the cluster analysis for six Aβ40 peptides, the snapshot structures were separated into four major and one fractional groups in the mixed solution. One of the four major groups consisted of the snapshots of one Aβ peptide labeled as “a” and was largely distant from other groups (Fig. S15a). Other groups were mixtures of the snapshots of two or three Aβ peptides. Therefore, the structural conversion of Aβ40 occurred more readily than that of Aβ42. In the aqueous solution of Aβ40, six groups were observed in the cluster analysis (Fig. S15b). Each of the major groups almost consisted of the snapshots of one specific Aβ, which suggested that the motion of Aβ40 was restricted in the Aβ assembly in the aqueous environment. It is confirmed from the dendrograms that the dissimilarities among the group members in the mixed solution are larger than those in the aqueous solution.
Fibrillation of Aβ42 and Aβ40According to the experiment on the Aβ fibrillation by Fukunaga et al.,14) Aβ40 developed into a fibril within several hours in the mixed solution of 80% 1,4-dioxane and 20% water. In contrast, the Aβ fibrillation in the aqueous solution required much longer incubation time. The Aβ40 fibrils formed in the mixed solution were morphologically similar to the toxic amyloid fibrils formed on the GM1 ganglioside-containing membrane. Measurement with the Fourier-transform (FT) IR spectroscopy indicated that the Aβ fibrils formed in the mixed membrane were β-sheet-rich structure. In our computational study, Aβ peptides were likely to expand its C-terminal domains outward in the mixed solution. The outward expansion increases the chances for the C-terminal domain to interact with another peptide. Hence, our simulation is compatible with the experimental results in the point that the mixed solution enhances the inter-molecular interaction among Aβ peptides, which will promote the fibril formation of Aβs. In the DSSP analysis, β-strand or β-bridge is not the major secondary structure. The fibril formation needed several hours in the mixed solution,14) while our simulation time was 5 µs at most. If the simulation is executed much longer, a seed for β-sheet may be observed. Although the residues involved in many inter-molecular hydrogen bonds showed the turn or bend in our simulation, the conformation of the residues is expected to be straight and close to the β-sheet form in the long-time simulation. In addition to the simulation time, the number of Aβ peptides in the calculation model is needed to be increased to raise the Aβ concentration for optimal calculation condition suitable for β-sheet formation. The number of Aβ molecules in a β-sheet-rich oligomer was estimated to be about 15 for fibrillation on GM1-containing artificial membranes30) and neuronal cells.31)
The commonly observed forms of toxic Aβ peptides consist of 40 and 42 residues, and the latter is the major constituent of senile plaque.32) An experimental study suggested that both Aβ40 and Aβ42 exhibited an affinity to the lipid membrane, but Aβ42 was more likely to form amyloid fibrils.33) Our simulation is compatible with these experimental results because Aβ42 conformation tends to be loose in the mixed solution remarkably compared to Aβ40.
Comparison with the Previous Calculation StudiesThe secondary structure of a single Aβ40 was computationally analyzed, changing the solution condition regarding pH and salt concentration.34) The difference in secondary structure was also examined between the normal Aβ40 and the Met35-oxidized one. The MD simulation study showed that solution pH and salt concentration had little influence on the normal Aβ40, which intimated the importance of another factor other than solution pH and ionic strength. The hydrophobicity focused in the present study is a factor not investigated in the previous study.
The interaction of two Aβ40 peptides was examined by the MD simulations with several kinds of lipid membranes with different lipid components.35) The simulation showed that a part of the Aβ peptide was expanded on the lipid membrane, which is consistent with our present study. MD simulations were also reported for the interaction of four Aβ42 peptides on the mixed lipid membrane.36) Aβ tetramer was initially built in the solution without membrane, and then the Aβ tetramer was placed on the membrane surface. A modulation in the structure of Aβ42 tetramer was observed, in which the Aβ42 tetramer was elongated in the presence of membrane, and the contact area among Aβ42 peptides was rearranged. The β-strand content was slightly increased on the raft-mimicking membrane model, and the C-terminal region of an Aβ peptide was loosened, which is consistent with our present simulation result.
An MD simulation for short amyloid peptide (Aβ13–23) was performed, including the effect of the lipid membrane by making a mixture of water and cyclohexane as a calculation model.37) The β-sheet formation emerged during the simulation, while the helix content was diminished at the late stage of the 0.5 µs simulation. Although the short peptide can readily change its conformation, the β-sheet was stably formed in the simulation with the hydrophobic environment.
The replica exchange molecular dynamics (REMD) simulation was adapted for Aβ42, Aβ40, and Met35-oxidized Aβ42.38) Anti-parallel β-hairpins were observed between Leu17–Ala21, Ala30–Leu36, and Val39–Ile41, which were expected to work as a seed for Aβ oligomer and fibril. The results intimated that the rate of the fibril formation would be low for Aβ40 and Met35-oxidized Aβ42, compared to Aβ42. The prediction of the fibrillation rate is compatible with our present simulation. The aggregation of four Aβ40 peptides was investigated by a combination of the fluorescence anisotropy technique with fluorescing Tyr side chains and the computer simulation with Monte Carlo (MC) and MD methods.39) In the aqueous solution, Aβ40 had a large amount of helix structure. Four Aβ monomers were bound to each other to make an assembly, which is compatible with our results in Fig. 1b.
An energy landscape for the Aβ fibrillation was suggested by a recent MD simulation study.40) An isolated monomer was in the disordered form with a short helix at the central hydrophobic core region (Leu17–Asp23). A hairpin structure, however, became stable in the oligomer because of the formation of hydrogen bonds between neighboring monomers. The prefibrillar oligomer mainly consisted of anti-parallel β-sheet, which would be converted into a fibrillar oligomer with parallel β-sheet. The energy change for this conversion was nearly downhill. In view of this energy landscape, the gathering of Aβ peptides shown in our present study is still at the initial stage of the fibrillation process, and the content of β-strand or β-bridge is still small.
Most of the previous computational studies on the interaction of multiple Aβ peptides were performed in the aqueous solution. For example, MD simulations of Aβ42 trimer and pentamer in water were reported for analyzing the stability of the inter-molecular contact.41) The trimer and pentamer were set to be in a hairpin shape with two β-sheet regions (residues 18–26 and 31–42), derived from an NMR structure (PDB#: 2BEG). The two β-sheet regions kept their conformations during the simulation in solution, while the residues 31–42 were more tightly bundled than the residues 18–26. This simulation suggested that the β-sheet structure of Aβ peptides was stable in the aqueous solution once the sheet formation was established.
Conclusion
Multiple MD simulations were performed to examine the conformations of Aβ42 and Aβ40 peptides and their complex structure in the hydrophobic environment. The hydrophobicity was provided by a mixture of 80% dioxane and 20% water in the calculation model. One, four, or six Aβ peptides were placed in the mixed solution and 1 µs simulations were performed for the respective models. For the six Aβs model, the simulations were extended to 5 µs. MD simulations were also performed in the aqueous solution without dioxane for comparison. It was demonstrated by the simulations that Aβ42 peptides gathered to be a single complex both in the mixed and aqueous solutions. In the mixed solution, the folding of the Aβ42 peptide was loose. The C-terminal hydrophobic domains were positioned outside of the Aβ complex and tended to spread outward. The N-terminal polar domains were positioned inside. In contrast, Aβ42 peptides were tightly packed in the aqueous solution. The simulation results for Aβ40 peptides were almost consistent with those of Aβ42. The C-terminal domains of Aβ40 stretched outward in the mixed solution, and stable inter-molecular interactions were observed at the C-terminal domains. In this simulation study, a markedly stable interaction was generated within 5 µs for Aβ42 peptides, in which two Aβs took a formation of the parallel β-sheet structure. The parallel β-sheet structure is expected to be a seed for the Aβ fibrils.
Acknowledgments
Calculations were performed at Research Center for Computational Science, Okazaki, Japan and Information Technology Center of the University of Tokyo. A part of this work was supported by a Grant for Scientific Research C from Japan Society for the Promotion of Science.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials. Conformational changes of six Aβ42 and six Aβ40 peptides during the 5 µs simulation; changes in the number of inter-molecular hydrogen bonds; frequencies of the secondary structures for the Aβ42 and six Aβ40 peptides; cluster analysis on the snapshot structures of six Aβ42 and six Aβ40 peptides for the last 1 µs.
References
- 1) LaFerla F. M., Green K. N., Oddo S., Nat. Rev. Neurosci., 8, 499–509 (2007).
- 2) Chiti F., Dobson C. M., Annu. Rev. Biochem., 75, 333–366 (2006).
- 3) Butterfield S. M., Lashuel H. A., Angew. Chem. Int. Ed., 49, 5628–5654 (2010).
- 4) Murphy R. M., Biochim. Biophys. Acta Biomembr., 2007, 1923–1934 (1768).
- 5) Sabaté R., Estelrich J., J. Phys. Chem. B, 109, 11027–11032 (2005).
- 6) Matsuzaki K., Biochim. Biophys. Acta Biomembr., 2007, 1935–1942 (1768).
- 7) Matsuzaki K., Kato K., Yanagisawa K., Biochim. Biophys. Acta, 2010, 868–877 (1801).
- 8) Yano Y., Takeno A., Matsuzaki K., Biochim. Biophys. Acta, 2018, 1603–1608 (1860).
- 9) Ariga T., Kobayashi K., Hasegawa A., Kiso M., Ishida H., Miyatake T., Arch. Biochem. Biophys., 388, 225–230 (2001).
- 10) Hayashi H., Kimura N., Yamaguchi H., Hasegawa K., Yokoseki T., Shibata M., Yamamoto N., Michikawa M., Yoshikawa Y., Terao K., Matsuzaki K., Lemere C. A., Selkoe D. J., Naiki H., Yanagisawa K., J. Neurosci., 24, 4894–4902 (2004).
- 11) Kakio A., Nishimoto S., Yanagisawa K., Kozutsumi Y., Matsuzaki K., J. Biol. Chem., 276, 24985–24990 (2001).
- 12) Kakio A., Nishimoto S., Yanagisawa K., Kozutsumi Y., Matsuzaki K., Biochemistry, 41, 7385–7390 (2002).
- 13) Okada T., Ikeda K., Wakabayashi M., Ogawa M., Matsuzaki K., J. Mol. Biol., 382, 1066–1074 (2008).
- 14) Fukunaga S., Ueno H., Yamaguchi T., Yano Y., Hoshino M., Matsuzaki K., Biochemistry, 51, 8125–8131 (2012).
- 15) Tomaselli S., Esposito V., Vangone P., van Nuland N. A., Bonvin A. M., Guerrini R., Tancredi T., Temussi P. A., Picone D., ChemBioChem, 7, 257–267 (2006).
- 16) Hoshino T., Mahmood M. I., Mori K., Matsuzaki K., J. Phys. Chem. B, 117, 8085–8094 (2013).
- 17) Vivekanandan S., Brender J. R., Lee S. Y., Ramamoorthy A., Biochem. Biophys. Res. Commun., 411, 312–316 (2011).
- 18) Vahed M., Neya S., Matsuzaki K., Hoshino T., J. Phys. Chem. B, 122, 3771–3781 (2018).
- 19) Case D., Betz R., Cerutti D. S., Cheatham T., Darden T., Duke R., Giese T. J., Gohlke H., Götz A., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., Lee T.-S., LeGrand S., Li P., Lin C., Luchko T., Kollman P., Amber 16, University of California, San Francisco, 2016.
- 20) Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., J. Chem. Theory Comput., 11, 3696–3713 (2015).
- 21) Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., J. Comput. Chem., 25, 1157–1174 (2004).
- 22) Qi F., Yoneda T., Neya S., Hoshino T., J. Phys. Chem. B, 122, 8503–8515 (2018).
- 23) Fudo S., Yamamoto N., Nukaga M., Odagiri T., Tashiro M., Hoshino T., Biochemistry, 55, 2646–2660 (2016).
- 24) The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC.
- 25) Qi F., Fudo S., Neya S., Hoshino T., J. Chem. Inf. Model., 55, 1673–1685 (2015).
- 26) Touw W. G., Baakman C., Black J., te Beek T. A. H., Krieger E., Joosten R. P., Vriend G., Nucleic Acids Res., 43 (D1), D364–D368 (2015).
- 27) Rochet J. C., Lansbury P. T. Jr., Curr. Opin. Struct. Biol., 10, 60–68 (2000).
- 28) Hou L., Kang I., Marchant R. E., Zagorski M. G., J. Biol. Chem., 277, 40173–40176 (2002).
- 29) Crescenzi O., Tomaselli S., Guerrini R., Salvadori S., D’Ursi A. M., Temussi P. A., Picone D., Eur. J. Biochem., 269, 5642–5648 (2002).
- 30) Ikeda K., Yamaguchi T., Fukunaga S., Hoshino M., Matsuzaki K., Biochemistry, 50, 6433–6440 (2011).
- 31) Itoh N., Takada E., Okubo K., Yano Y., Hoshino M., Sasaki A., Kinjo M., Matsuzaki K., ChemBioChem, 19, 430–433 (2018).
- 32) Török M., Milton S., Kayed R., Wu P., McIntire T., Glabe C. G., Langen R., J. Biol. Chem., 277, 40810–40815 (2002).
- 33) Ogawa M., Tsukuda M., Yamaguchi T., Ikeda K., Okada T., Yano Y., Hoshino M., Matsuzaki K., J. Neurochem., 116, 851–857 (2011).
- 34) Brown A. M., Lemkul J. A., Schaum N., Bevan D. R., Arch. Biochem. Biophys., 545, 44–52 (2014).
- 35) Lemkul J. A., Bevan D. R., Biochemistry, 52, 4971–4980 (2013).
- 36) Brown A. M., Bevan D. R., Biophys. J., 111, 937–949 (2016).
- 37) Bajda M., Filipek S., Comput. Biol. Chem., 56, 13–18 (2015).
- 38) Rosenman D. J., Connors C. R., Chen W., Wang C., García A. E., J. Mol. Biol., 425, 3338–3359 (2013).
- 39) Mancini O., Wellbrock T., Rolinski O. J., Kubiak-Ossowska K., Mulheran P. A., Phys. Chem. Chem. Phys., 20, 4216–4225 (2018).
- 40) Zheng W., Tsai M. Y., Chen M., Wolynes P. G., Proc. Natl. Acad. Sci. U.S.A., 113, 11835–11840 (2016).
- 41) Masman M. F., Eisel U. L. M., Csizmadia I. G., Penke B., Enriz R. D., Marrink S. J., Luiten P. G. M., J. Phys. Chem. B, 113, 11710–11719 (2009).