Introduction
Weinreb amides1–3) are well known as the synthetic equivalents of aldehydes and ketones and are commonly used in synthetic organic chemistry. Therefore, the selective conversions of α,β-unsaturated Weinreb amides in the presence of similarly reactive α,β-unsaturated esters provide a useful approach. However, no such conversion has been reported. As an extension of our efforts to develop in situ protection methods for carbonyl derivatives,4–11) we attempted to apply this method to a combination of α,β-unsaturated Weinreb amides and α,β-unsaturated esters.
First, the relative reactivities of α,β-unsaturated ester 1 and α,β-unsaturated Weinreb amide 2 were investigated (Chart 1). That is, 1 equivalent (equiv) of diisobutylaluminium hydride (DIBAL-H) (the same mole as 1) was added to a 1 : 1 mixture of 1 and 2 in CH2Cl2 at −78 °C, and the mixture was stirred for 3 h at the same temperature. As a result, 7% of α,β-unsaturated aldehyde 3, 24% of ally alcohol 4, and 33% of α,β-unsaturated aldehyde 5 were obtained along with 56% of recovered 1 and 61% of 2. This result indicated that selective reduction of one in the presence of the other was difficult, although the reactivity of the α,β-unsaturated Weinreb amide was slightly lower than that of the α,β-unsaturated ester.

Chart 1. Comparison of the Reactivity of α,β-Unsaturated Ester 1 and α,β-Unsaturated Weinreb Amide 2 in DIBAL-H Reduction
Next, an in situ protection method was investigated in hope of salt formation from a slightly reactive α,β-unsaturated ester. However, the usual specific conditions for unsaturated systems, PPh3 and trimethylsilyl trifluoromethanesulfonate (TMSOTf), did not work well, and little salt was formed. This may have been due to the electron-donating effect of the alkoxy oxygen atom and the relatively weak nucleophilicity of PPh3.
We then reviewed the conditions for the formation of salts from α,β-unsaturated esters and focused mainly on investigating phosphines more electron-rich than PPh3 to enhance nucleophilicity (Table 1). The α,β-unsaturated ester 1, PR3 (1.5 equiv) and TMSOTf (1.5 equiv) in CH2Cl2 were refluxed for 48 h. As a result, P(C6H4-p-Me)3 (Entry 3), P(C6H4-p-OMe)3 (Entry 5), and trialkyl phosphines such as PEt3 and P(n-Bu)3 (Entries 6 and 7) formed phosphonium salts. Salt formation could be easily detected by TLC, as highly polar compound appeared after the disappearance of the starting ester 1. In particular, P(C6H4-p-OMe)3 and PEt3 formed salts in a short time (Entries 5 and 7). Considering the price point, the following survey was conducted using PEt3.
Table 1. Investigation of Phosphines in Phosphonium Salt Formation
a) 72 h were required for salt formation. b) After 24 h, salt was generated. c) Figure below shows TLC behavior of the reaction in the case of Entry 5.

In addition, the use of the higher boiling point toluene instead of CH2Cl2 reduced the time. That is, α,β-unsaturated ester 1, PEt3 (1.5 equiv) and TMSOTf (1.5 equiv) in toluene were refluxed for 5 h to form the phosphonium ion (Chart 2).

Chart 2. Phosphonium Ion Formation in Toluene
The optimized conditions were then used to compare the reactivities of unsaturated esters and unsaturated Weinreb amides toward nucleophiles (Chart 3). To a 1 : 1 mixture of α,β-unsaturated ester 1 and α,β-unsaturated Weinreb amide 2 in toluene were added, dropwise, PEt3 (1.5 equiv based on the number of moles of 1) and TMSOTf (1.5 equiv) at room temperature (r.t.). The resulting solution was stirred under reflux for 5 h. Subsequently, the solvent was cooled to −78 °C, and DIBAL-H (2.0 equiv) was added dropwise. After consuming 2, the mixture was treated with tetrabutylammonium fluoride (TBAF). As a result, 90% of α,β-unsaturated aldehyde 5 was obtained from Weinreb amide 2 along with 91% of recovered 1. From the above results, it was suggested that the salt was formed selectively from the α,β-unsaturated ester 1.
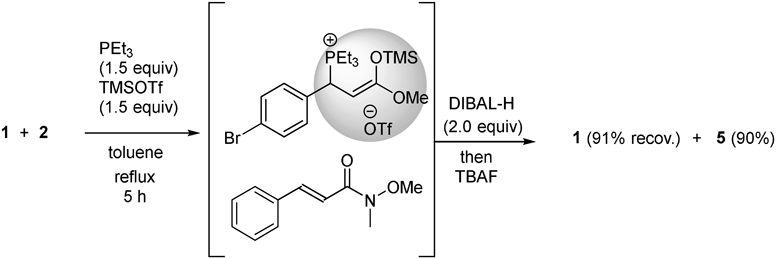
Chart 3. Reduction of the 1 : 1 Mixture of α,β-Unsaturated Ester 1 and α,β-Unsaturated Weinreb Amide 2 with DIBAL-H in Toluene with In Situ Protection
Next, the generality of substrates for the selective reduction reaction of α,β-unsaturated Weinreb amides in the presence of α,β-unsaturated esters was examined (Table 2). In this reaction, not only α,β-unsaturated ester 1 with an aromatic ring at the β-position, but also α,β-unsaturated ester 6 with aliphatic chains (Entries 1–4, 6) and a cyclic α,β-unsaturated ester, coumarin (9) (Entry 5), were available to form selectively the corresponding phosphonium salts. The coexisting α,β-unsaturated Weinreb amides having an aromatic ring (2) and an aliphatic chain (7) at the β-position were reduced selectively to give the corresponding α,β-unsaturated aldehydes in good yields (Entries 1–5). Furthermore, after salt formation, a methyl group could be introduced into Weinreb amide 2 by using a Grignard reagent instead of DIBAL-H to afford the α,β-unsaturated ketone 10 (Entry 6).
Table 2. Selective Reduction of α,β-Unsaturated Weinreb Amides in the Presence of α,β-Unsaturated Esters
a) TESOTf was used instead of TMSOTf. b) MeMgBr (2.0 equiv) was used instead of DIBAL-H.
In conclusion, we have developed an easy, selective conversion of α,β-unsaturated Weinreb amides in the presence of α,β-unsaturated esters. To achieve this selective conversion, new conditions for in situ protection have been developed. Since Weinreb amides are synthetic equivalents of aldehydes and ketones, the method here would provide a useful tool in synthetic organic chemistry.
Experimental
General InformationAll reagents were purchased from commercial sources. Reactions were performed under a nitrogen atmosphere using purchased anhydrous solvent. All reactions were monitored by TLC using Merck silica gel 60 F254. The products were purified by column chromatography over silica gel Kieselgel 60 (70–230 mesh ASTM) purchased from Merck or Silica Gel 60N (40–50 µm, spherical neutral) purchased from Kanto Chemical (Tokyo, Japan). 1H-NMR and 13C-NMR spectra were recorded at 25 °C on a JEOL JNM-AL300 (at 300 MHz and 75 MHz, respectively), a JEOL JNM-ECS 400 (at 400 MHz and 100 MHz, respectively) or a JEOL JNM-LA 500 (at 500 MHz and 125 MHz, respectively), and the chemical shifts are reported relative to internal tetramethylsilane (TMS) (1H, δ = 0.00) and CDCl3 (13C, δ = 77.0). Data for 1H-NMR spectra are reported as follows: chemical shift (δ ppm) (integration, multiplicity, coupling constant (Hz)). Multiplicity and qualifier abbreviations are as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. IR spectra (KBr) were recorded by an SHIMADZU FTIR-8400 or SHIMADZU IRAffinity-1, and are reported in frequivuency of absorption (cm−1). High-resolution mass spectra (MALDI-TOF) were performed by the Elemental Analysis Section of Graduate School of Pharmaceutical Science in Osaka University.
The substrates 1, 3, 6, 9 are commercially available.
Weinreb amide 212): To a solution of benzaldehyde (1.0 g, 9.4 mmol) in CH2Cl2 (19 mL, 0.50 M) was slowly added N-methoxy-N-methyl-2-(triphenylphosphoranylidene) acetamide (4.1 g, 11.3 mmol, 1.2 equiv) at r.t. After being stirred for 5 h at r.t., the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (n-hexane/AcOEt = 1/1) to afford 2 (1.7 g, 9.0 mmol, 96%) as a colorless oil. 1H-NMR (500 MHz, CDCl3) δ: 7.73 (1H, d, J = 15.7 Hz), 7.55 (2H, m), 7.35 (3H, m), 7.04 (1H, d, J = 15.7 Hz), 3.74 (3H, s), 3.29 (3H, s).
Weinreb amide 713): To a solution of decanal (1.0 g, 6.4 mmol) in CH2Cl2 (13 mL, 0.50 M) was slowly added N-methoxy-N-methyl-2-(triphenylphosphoranylidene) acetamide (2.8 g, 7.7 mmol, 1.2 equiv) at r.t. After being stirred for 5 h at r.t., the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (n-hexane/AcOEt = 1/1) to afford 7 (1.4 g, 5.8 mmol, 90%) as a colorless oil. 1H-NMR (500 MHz, CDCl3) δ: 6.97 (1H, dt, J = 15.7, 5.0 Hz), 6.38 (1H, d, J = 15.7 Hz), 3.70 (3H, s), 3.23 (3H, s), 2.22 (2H, dt, J = 5.0, 1.6 Hz), 1.46 (2H, m) 1.29 (12H, m), 0.88 (3H, m).
Experimental Details for Chart 1A solution of 1 (240.0 mg, 1.00 mmol) and 2 (191.1 mg, 1.00 mmol) in CH2Cl2 (10 mL, 0.1 M) was cooled to −78 °C. DIBAL-H (1.0 M toluene solution, 1.0 mL, 1.0 equiv) was added dropwise to the reaction mixture, and the reaction mixture was stirred for 2 h. After the reaction mixture was quenched with 1N HCl and the solvent volume was reduced under vacuum. The residue left behind was extracted with ethyl acetate (EtOAc) (3 × 30 mL). The organic layer was separated and dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (n-hexane/EtOAc = 2/1) to afford the recovered 1 (134.4 mg, 0.56 mmol, 56%) and 2 (116.6 mg, 0.61 mmol, 61%), and the reduced products 3 (14.7 mg, 0.07 mmol, 7%), 4 (50.9 mg, 0.24 mmol, 24%) and 5 (43.6 mg, 0.33 mmol, 33%).
3: 1H-NMR (400 MHz, CDCl3) δ: 9.69 (1H, d, J = 7.4 Hz), 7.57 (2H, d, J = 6.4 Hz), 7.42 (2H, d, J = 6.4 Hz), 7.41 (1H, d, J = 16.0 Hz), 6.69 (1H, dd, J = 16.0, 7.4 Hz).
414): 1H-NMR (300 MHz, CDCl3) δ: 7.44 (2H, d, J = 8.6 Hz), 7.24 (2H, d, J = 8.6 Hz), 6.56 (1H, d, J = 15.9 Hz), 6.35 (1H, dt, J = 15.9, 5.6 Hz), 4.32 (2H, d, J = 5.6 Hz), 1.59 (1H, brs, OH).
Experimental Details for Chart 3, Table 2General procedure: To a solution of α,β-unsaturated Weinreb amide (1.00 mmol, 1.0 equiv), α,β-unsaturated ester (1.00 mmol, 1.0 equiv) and PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv) in toluene (10 mL) was added dropwise TMSOTf (272 µL, 1.50 mmol, 1.5 equiv) at r.t. After being stirred for 5 h at 110 °C, the reaction mixture was then cooled to −78 °C. DIBAL-H (2.0 mL, 1.0 M n-hexane solution, 2.0 equiv) was added to the reaction mixture. After the starting α,β-unsaturated Weinreb amide was consumed, suspension of TBAF (1.0 M in THF, 3.0 mL, 3.0 equiv) was added, then the resulting solution was stirred for 30 min. After adding H2O, the mixture was extracted with CH2Cl2. The extract was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography to afford the desired products.
Cinnamaldehyde (5)15) (Charts 1 and 3, Table 2, Entries 1, 2, and 5)Entry 1: According to the general procedure, 1 (240.0 mg, 1.00 mmol), 2 (191.1 mg, 1.00 mmol), PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv), TMSOTf (272 µL, 1.50 mmol, 1.5 equiv), DIBAL-H (2.0 mL, 1.0 M n-hexane solution, 2.0 equiv), and suspension of TBAF (1.0 M, 3.0 mL, 3.0 equiv) gave recovered 1 (218.4 mg, 0.91 mmol, 91%) and 5 (118.9 mg, 0.90 mmol, 90%) as a colorless oil after purification by flash column chromatography (n-hexane/AcOEt = 6/1).
Entry 2: According to the general procedure, 6 (156.1 mg, 1.00 mmol), 2 (191.1 mg, 1.00 mmol), PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv), TMSOTf (272 µL, 1.50 mmol, 1.5 equiv), DIBAL-H (2.0 mL, 1.0 M n-hexane solution, 2.0 equiv), and suspension of TBAF (1.0 M, 3.0 mL, 3.0 equiv) gave recovered 6 (139.0 mg, 0.89 mmol, 89%) and 5 (121.5 mg, 0.92 mmol, 92%) as a colorless oil after purification by flash column chromatography (n-hexane/AcOEt = 6/1).
Entry 5: According to the general procedure, 9 (146.0 mg, 1.00 mmol), 2 (191.1 mg, 1.00 mmol), PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv), TMSOTf (272 µL, 1.50 mmol, 1.5 equiv), DIBAL-H (2.0 mL, 1.0 M n-hexane solution, 2.0 equiv), and suspension of TBAF (1.0 M, 3.0 mL, 3.0 equiv) gave recovered 9 (141.6 mg, 0.97 mmol, 97%) and 5 (116.3 mg, 0.88 mmol, 88%) as a colorless oil after purification by flash column chromatography (n-hexane/AcOEt = 3/1).
1H-NMR (400 MHz, CDCl3): δ = 9.70 (1H, d, J = 7.6 Hz), 7.56 (2H, m), 7.50 (m, 1H), 7.46–7.44 (m, 3H), 6.75–6.70 (m, 1H).
(E)-Dodec-2-enal (8)16) (Table 2, Entries 3 and 4)Entry 3: According to the general procedure, 1 (240.0 mg, 1.00 mmol), 7 (241.2 mg, 1.00 mmol), PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv), TMSOTf (272 µL, 1.50 mmol, 1.5 equiv), DIBAL-H (2.0 mL, 1.0 M n-hexane solution, 2.0 equiv), and suspension of TBAF (1.0 M, 3.0 mL, 3.0 equiv) gave recovered 1 (216.0 mg, 0.90 mmol, 90%) and 8 (153.0 mg, 0.84 mmol, 84%) as a colorless oil after purification by flash column chromatography (n-hexane/AcOEt = 6/1).
Entry 4: According to the general procedure, 6 (156.1 mg, 1.00 mmol), 7 (241.2 mg, 1.00 mmol), PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv), TMSOTf (272 µL, 1.50 mmol, 1.5 equiv), DIBAL-H (2.0 mL, 1.0 M n-hexane solution, 2.0 equiv), and suspension of TBAF (1.0 M, 3.0 mL, 3.0 equiv) gave recovered 6 (132.7 mg, 0.85 mmol, 85%) and 8 (156.7 mg, 0.86 mmol, 86%) as a colorless oil after purification by flash column chromatography (n-hexane/AcOEt = 6/1).
1H-NMR (400 MHz, CDCl3): δ = 9.43 (1H, d, J = 7.5 Hz), 6.68 (1H, dt, J = 15, 7.5 Hz), 6.04 (1H, dd, J = 15, 7.5 Hz), 2.49–2.12 (2H, m), 2.02–1.06 (14H, m), 1.01–0.73 (3H, m).
(E)-4-Phenylbut-3-en-2-one (10)9) (Table 2, Entry 6)Entry 6: According to the general procedure, 1 (240.0 mg, 1.00 mmol), 2 (191.1 mg, 1.00 mmol), PEt3 (1.0 M in toluene solution, 1.5 mL, 1.50 mmol, 1.5 equiv), TESOTf (389 µL, 1.50 mmol, 1.5 equiv), MeMgBr (2.0 mL, 1.0 M THF solution, 2.0 equiv), and suspension of TBAF (1.0 M, 3.0 mL, 3.0 equiv) gave recovered 1 (199.2 mg, 0.83 mmol, 83%) and 10 (119.8 mg, 0.82 mmol, 82%) as a colorless oil after purification by flash column chromatography (n-hexane/AcOEt = 7/1).
1H-NMR (500 MHz, CDCl3) δ: 7.58–7.48 (m, 3H), 7.43–7.37 (3H, m), 6.71 (1H, d, J = 16.1 Hz), 2.39 (s, 3H).

