Notes
Computational Study on the Synergic Effect of Brønsted Acid and Hydrogen-Bonding Catalysis for the Dearomatization Reaction of Phenols with Diazo Functionality
2020 Volume 68 Issue 11 Pages 1104-1108
Details
2020 Volume 68 Issue 11 Pages 1104-1108
Catalytic dearomative transformations of phenol variants via an ipso-Friedel-Crafts reaction could provide a straightforward method for the rapid assembly of functionalized spiromolecules as versatile synthetic scaffolds. We previously reported a dearomative spirocyclization reaction by merging Brønsted acid and hydrogen-bonding catalysis. However, it was unclear how the reaction proceeded and how the synergic effect was triggered. Described herein are the computational studies used to elucidate the reaction mechanism. Such calculations indicated that the applied catalysts, maleic acid and Schreiner’s thiourea, work cooperatively. The synergic effect enabled the chemoselectivity to interconvert between phenol dearomatization and O–H insertion, which is a major side reaction. This investigation also revealed that not only does the Schreiner’s thiourea catalyst serve as a hydrogen bonding donor, but the sulfur atom in thiourea possesses a general base function. The dual functional support of the thiourea along with maleic acid would thus realize the chemoselective prioritization of dearomatization over the O–H insertion reaction under mild conditions.
Merging two distinct catalysts often enhances the chemical yield and selectivity, or enables challenging reactions that cannot be realized by employing a single catalyst system.1,2) Recent achievements using anion-binding thiourea or its variants as widely available co-catalysts in a cooperative catalyst system have begun to gain momentum.3–16) In 2016, Hayashi and Ogasawara developed a multiple catalyst system comprising Schreiner’s thiourea to accelerate the reaction and enhance the yield and selectivities of a time and pot economical total synthesis of (−)-oseltamivir.17) In 2018, Scheidt and colleagues reported the asymmetric synthesis of indole-fused tetrahydropyrans using a cooperative catalysis system.18) They proposed that an achiral urea, as a hydrogen bond donor, chiral phosphoric acid, and the substrate interacted to form a key transition state that enhanced the yield, selectivity, and reaction rate. These kinds of cooperative catalysis systems have been applied to diverse reaction systems.19) However, a detailed reaction mechanism remains unclear for many cases.
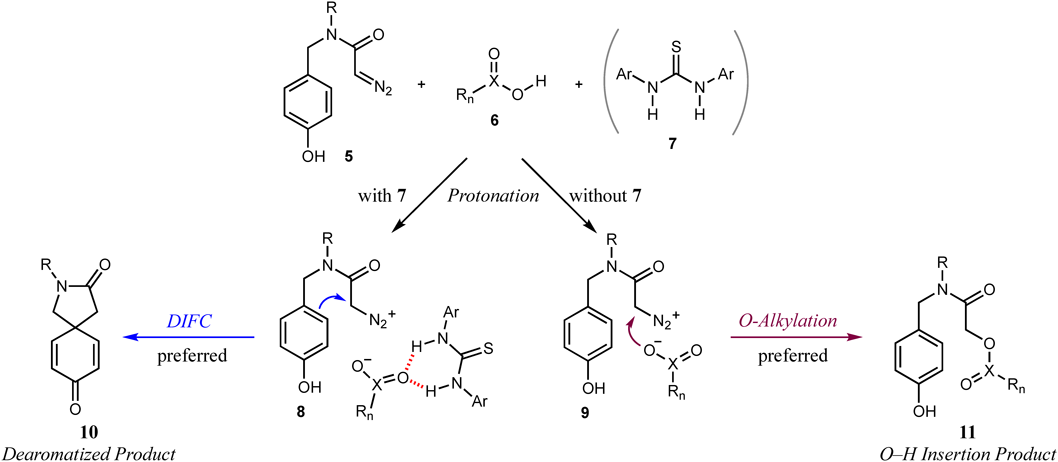
In our continuous research program to develop dearomative spirocyclizations20,21) and to investigate metal-carbene chemistry using diazo compounds,22–29) we reported a phenol dearomatization utilizing a cooperative catalyst system consisting of 10 mol% maleic acid and Schreiner’s thiourea in 2018.30) This methodology provides rapid access to functionalized spirolactams, which are ubiquitous pivotal architectures in biologically useful natural products and pharmaceuticals31,32) (Fig. 1). The reaction without maleic acid 3 did not produce target compound 2, and a low conversion was observed when using only 3 without Schreiner’s thiourea 4 (Chart 1). The metal-free conditions in the reaction using diazo compounds contributed to the excellent chemoselectivity by suppressing Büchner ring expansion and C–H insertion, which are specific to the metal-carbene reaction.

(Color figure can be accessed in the online version.)

Preliminary experimental studies to shed light on the reaction process were performed. Such studies indicated that O–H insertion with a Brønsted acid catalyst occurred as a side reaction under the conditions without Schreiner’s thiourea.33) Therefore, we proposed a plausible reaction pathway, as shown in Chart 2. The two reactions would proceed through a stepwise mechanism comprising the protonation of a diazo group by a Brønsted acid catalyst followed by the nucleophilic substitution reaction of the generated carboxylate anion or phenol unit. However, the exact mechanism remained unclear, and computational supporting evidence were not provided. In this paper, we describe the computational studies to elucidate the origin of the chemoselectivity inversion between the phenol dearomatization and O–H insertion reactions.
(Color figure can be accessed in the online version.)
Quantum chemical computations on the basis of density functional theory (DFT) calculations were employed to illustrate the reaction coordinate diagrams for dearomative spirocyclization and the O–H insertion reaction.34) All the calculations were performed at the Rwb97xd/6-311++G(d,p)//Rb3lyp/6-31 + g(d) level of theory (in MeCN solvent). The intrinsic reaction coordinate (IRC) method was used to track the minimum energy paths from the transition structures to the corresponding local minima.35) First, we analyzed the protonation event of the diazo functionality using maleic acid as a Brønsted acid. The results indicated that a slightly endothermic formation of INT1 from SM occurred (ΔG=+2.49 kcal/mol, Fig. 2). In INT1, C–H…O non-classical hydrogen bonding interactions were present between maleic acid and the benzylic C–H bonds (2.61 Å).36) Generation of diazonium salt INT2 proceeded with a reasonable activation energy of ΔG‡=+12.39 kcal/mol via TS1, which possesses an O–H…O classical hydrogen bonding interaction between the phenolic hydroxyl group and the catalyst (2.14 Å).

(Color figure can be accessed in the online version.)
Figure 3 shows the reaction coordinate diagrams of the DIFC reaction and O–H insertion reaction starting from INT2 in the absence of a thiourea catalyst. The transition structures of these processes (TSDIFC and TSO−H, respectively) had similar activation energies (ΔG‡=+19.71 kcal/mol and 19.30 kcal/mol, respectively). However, the O–H insertion reaction was kinetically favored slightly, based on the energy profile (ΔΔG‡=+0.41 kcal/mol). Although the dearomatized product PRODIFC was calculated to be more thermodynamically stable than the O–H insertion product PROO−H (ΔΔG=−9.59 kcal/mol), such stability would be not critical for the product determination as these reactions are not reversible processes.

(Color figure can be accessed in the online version.)
We then investigated the reactions with a thiourea catalyst to understand the interconversion of the chemoselectivity (Fig. 4). To reduce the computational time-cost, structurally simplified 1,3-bis(3,5-difluorophenyl)thiourea was used. Intriguingly, the DIFC reaction proceeded through a 15-membered cyclophane-like transition structure TSDIFC-UREA via hydrogen bonding interactions between the dual catalysts and the substrate (ArO–H…S–C: 2.35 Å),37) thus affording the kinetically preferred product PRODIFC-UREA. TSO−H-UREA also seemed to have hydrogen bonding interactions between the phenolic hydroxyl group and the thiourea catalyst. However, in this case, the phenol component was not a nucleophilic species for the O–H insertion reaction. In addition, twin interactions between thiourea and maleic acid (1.96 and 1.98 Å) in TSO−H-UREA led to the deactivation of carboxylate as the nucleophile by decreasing the electron density, thereby suppressing O–H insertion. These aspects could account for the significant distinction between the activation energies expressed by the following equations: ΔΔG‡DIFC = ΔG‡(TSDIFC-UREA) − ΔG‡(TSDIFC)=−2.92 kcal/mol, whereas ΔΔG‡O−H = ΔG‡(TSO−H-UREA) − ΔG‡(TSO−H)=−1.94 kcal/mol. The inherent energy balance of the conditions with/without thiourea in the dearomatization and O−H insertion reactions can therefore be expressed by the following equation: ΔΔΔG‡ = ΔΔG‡DIFC−ΔΔG‡O−H=−0.983 kcal/mol. These factors influenced the prioritization of the operative reaction pathway for the inversion of the chemoselectivity, thereby supporting the experimental results.

Ar represents 3,5-difluorophenyl group. (Color figure can be accessed in the online version.)
In conclusion, the reaction pathways for the dearomative spirocyclization and O−H insertion reactions of a phenol with a diazo functionality using maleic acid with/without a thiourea co-catalyst were delineated on the basis of DFT calculations. In all transition structures, including that of the protonation of the diazo group, multiple classical/non-classical hydrogen bonding interactions were present. However, these mutual interactions do not necessarily help the reaction proceed more effectively. In general, hydrogen bonding is acknowledged as a positive interaction. However, in the case of the reaction with a thiourea co-catalyst, hydrogen bonding interfered with the O−H insertion reaction pathway, thus leading to the chemoselectivity prioritizing phenol dearomatization. We anticipate that the mechanistic profiles of the cooperative catalyst systems would build a foundation for further research into designing novel reactions and improved catalysts.
All DFT calculations were performed with Gaussian 16 program and the ball and stick models were drawn with Avogadro software. The molecular structure optimizations were carried out using the hybrid density functional method based on Becke’s three-parameter exchange function and the Lee-Yang-Parr nonlocal correlation functional (rB3LYP)38) and the 6-31 + G* basis set for H, C, N, O and S in MeCN solvent. The vibrational frequencies were computed at the same level to check whether each optimized structure is an energy minimum (no imaginary frequency) or a transition state (one imaginary frequency) and to evaluate its zero-point vibrational energy (ZPVE) and thermal corrections at 298.15 K. Single point energies were calculated at the RwB97XD level39) using 6-311++G** basis set for H, C, N, O and S in MeCN solvent.
This work was supported by the Toray Science Foundation, The Naito Foundation, Takeda Science Foundation, Futaba Electronics Memorial Foundation, Chugai Award in Synthetic Organic Chemistry, Japan, JSPS KAKENHI (Grant Numbers JP18H02550, and 18H02550). Numerical calculations were carried out on SR24000 at the Institute of Management and Information Technologies, Chiba University of Japan.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.