Experimental
GeneralAll melting points were determined on Yanagimoto micro melting point apparatus and are uncorrected. IR spectra were recorded on Horiba IR-710. 1H-NMR spectra were recorded on a JEOL JNM ECA600 (600 MHz) or a JEOL JNM ECS400 (400 MHz) spectrometer at room temperature (r.t.); chemical shifts (δ) are reported in parts per million relative to tetramethylsilane. Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet; sept, septet; m, multiplet; br, broad. 13C-NMR spectra were recorded on a JEOL JNM ECA600 (150 MHz) or a JEOL JNM ECS400 (100 MHz) spectrometer with complete proton decoupling. chemical shifts are reported in parts per million relative to tetramethylsilane with the solvent resonance as the internal standard CDCl3. High resolution (HR)MS were recorded on a JEOL JMS-T100TD. X-ray crystallographic analysis was performed on Rigaku R-AXIS RAPIDII-S. Analytical TLC was performed on Merck precoated TLC plates (silica gel 60 GF254, 0.25 mm). Silica gel column chromatography was carried out on silica gel 60N (Kanto Kagaku Co., Ltd., spherical neutral, 63–210 or 40–50 µm). Preparative thin-layer chromatography (PTLC) was carried out on silica gel Wakogel B-5F. All reactions were carried out under nitrogen in dried glassware with magnetic stirring. Microwave experiments were carried out in sealed vessels in a synthesis reactor (Biotage Initiator 2.5). Compounds 6b,c were prepared by the reported method.34)
Synthesis of 2-(2-Vinylphenyl)phenol (6d)35)To a cooled suspension of methyltriphenylphosphonium iodide (128 mg, 0.318 mmol) in tetrahydrofuran (THF) (3 mL) at 0 °C was added potassium tert-butoxide (56 mg, 0.50 mmol) under nitrogen. After 30 min, 6H-benzo[c]chromen-6-one36) (25 mg, 0.15 mmol) which was prepared from phenyl 2-bromobenzoate was added. The reaction was warmed to r.t. and the mixture was stirred for 15 h. After dropping 1 N HCl until pH below 7, the aqueous phase was separated and extracted with DCM. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (Hexane/EtOAc) to give the product 6d (20 mg, 0.12 mmol, 81%). Compound 6d: 1H-NMR (CDCl3, 400 MHz) δ: 7.72 (1H, d, J = 7.2 Hz), 7.49–7.20 (4H, m), 7.13 (1H, d, J = 6.8 Hz), 6.99 (2H, d, J = 6.8 Hz), 6.56 (1H, dd, J = 17.6, 11.2 Hz), 5.75 (1H, d, J = 17.6 Hz), 5.22 (1H, d, J = 11.2 Hz), 4.80 (1H, s); 13C-NMR (CDCl3, 100 MHz) δ: 152.6, 136.7, 134.9, 134.4, 130.9, 130.7, 129.4, 128.6, 128.4, 126.7, 125.6, 120.5, 115.9, 115.5.
Synthesis of 2-(2-Vinylbenzyl)-1-(tert-butyldimethylsilyloxy)benzene (6e)A mixture of 1-bromomethyl-2-tert-butyldimethylsilyloxybenzene (1.38 g, 4.59 mmol), 2-formylphenylboronic acid (688 mg, 4.59 mmol), Pd(PPh3)4 (133 mg, 0.115 mmol), K2CO3 (1.90 g, 13.8 mmol) in THF (15 mL) was heated at 95 °C for 12 h. After cooling to room temperature, he reaction mixture was diluted by adding H2O and ethyl acetate. The mixture was extracted with ethyl acetate (three times), and the combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel (hexanes/ethyl acetate = 10 : 1) to afford 2-(2-((tert-butyldimethylsilyl)oxy)benzyl)benzaldehyde (29) (1.20 g, 3.68 mmol, 80%). Compound 29: 1H-NMR (CDCl3, 600 MHz) δ: 10.26 (1H, s), 7.88 (1H, d, J = 7.6 Hz), 7.49 (1H, t, J = 6.8 Hz), 7.39 (1H, t, J = 7.2 Hz), 7.15–7.10 (2H, m), 6.85–6.83 (3H, m), 4.00 (2H, s), 0.92 (9H, s), 0.23 (6H, s); 13C-NMR (CDCl3, 150 MHz) δ: 192.4, 153.3, 143.2, 134.0, 133.8, 131.1, 131.0, 130.7, 130.5, 127.4, 126.7, 121.2, 118.3, 32.6, 25.7, 18.2, −4.2; IR (CHCl3, cm−1): 2958, 2931, 2860, 1693; HRMS (DART+): m/z [M + H]+ Calcd for C20H26O2Si: 327.17803. Found: 327.17867.
To a stirred mixture of methyltriphenylphosphonium iodide (1.95 g, 4.82 mmol), t-BuOK (800 mg, 7.13 mmol) in THF (50 mL) was added a solution of 29 (750 mg, 2.30 mmol) in THF (30 mL) at 0 °C for 4 h. The reaction was quenched by addition of a solution of hydrochloric acid (<pH 7.0), and the mixture was extracted with ethyl acetate. The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel (hexanes) to afford the corresponding Wittig product (528 mg, 1.63 mmol, 71%).
To a stirred solution of the Wittig product (500 mg, 1.54 mmol) in THF (10 mL) was added a solution of TBAF in THF (1.0 M, 2.3 mL, 2.3 mmol) at 0 °C, and the reaction mixture was stirred at 0 °C for 25 min. The reaction was quenched by adding a solution of saturated aqueous NH4Cl. The resulting mixture was extracted with ethyl acetate (three times), and the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography on silica gel (hexanes/ethyl acetate = 10 : 1) to afford 6e (364 mg, 1.17 mmol, 76%). Compound 6e: 1H-NMR (CDCl3, 600 MHz) δ: 7.42 (1H, d, J = 7.2 Hz), 7.15–7.07 (2H, m), 7.01–6.95 (2H, m), 6.86 (1H, dd, J = 17.4, 11.4 Hz), 6.78 (1H, d, J = 7.2 Hz), 6.73 (1H, t, J = 7.8 Hz), 6.65 (1H, d, J = 8.4 Hz), 5.53 (1H, d, J = 17.4 Hz), 5.15 (1H, d, J = 11.4 Hz), 4.73 (1H, s), 3.92 (2H, s); 13C-NMR (CDCl3, 150 MHz) δ: 153.4, 137.1, 136.6, 134.5, 130.5, 129.8, 128.0, 127.6, 126.8, 126.5, 126.0, 120.9, 116.0, 115.3, 33.0; IR (CHCl3, cm−1): 3597, 3010; HRMS (DART+): m/z [M + H]+ Calcd for C14H13O1: 197.09664. Found: 197.09670.
The IMDA Reaction of 6d to 8d (Table 1, Entry 4)To a solution of 6d (68 mg, 0.35 mmol) in AcOH (3 mL) was added Pb(OAc)4 (922 mg, 2.08 mmol) at 0 °C, and the reaction temperature was allowed to reach room temperature. After 72 h, the precipitate was filtered off with EtOAc, and the filtrate was washed with saturated aqueous solution of NaHCO3. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/Hexane) to yield 8d (21 mg, 0.08 mmol, 23%) as a yellow solid. Compound 8d: mp: 128.0–129.0 °C (recryst. from CH2Cl2); 1H-NMR (CDCl3, 600 MHz) δ: 7.26–7.18 (4H, m), 6.59 (1H, t, J = 6.0 Hz), 6.39 (1H, t, J = 6.6 Hz), 4.38–4.35 (1H, m), 3.42–4.39 (1H, m), 3.27–3.22 (1H, m), 2.16 (3H, s), 1.89–1.88 (2H, m); 13C-NMR (CDCl3, 150 MHz) δ: 202.3, 169.2, 146.2, 139.3, 132.3, 131.5, 129.0, 127.2, 123.6, 122.6, 85.5, 52.1, 51.0, 38.4, 37.6, 21.5; IR (CHCl3, cm−1): 2951, 1738; HRMS (DART+): m/z [M + H]+ Calcd for C16H15O3: 255.10212. Found: 255.10187.
6-Acetoxy-6-(2-vinylbenzyl)cyclohexa-2,4-dien-1-one (7e)To a solution of 6e (54 mg, 0.24 mmol) in AcOH (1 mL) was added Pb(OAc)4 (317 mg, 0.715 mmol) at 0 °C, and the reaction temperature was allowed to reach room temperature. After 1 h, the precipitate was filtered off with EtOAc, and the filtrate was washed with saturated aqueous solution of NaHCO3. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/Hexane/1% triethylammonium acetate (TEA)) to yield 7e (15 mg, 0.055 mmol, 23%) as a yellow oil. Compound 7e: 1H-NMR (CDCl3, 600 MHz) δ: 7.52 (1H, d, J = 7.8 Hz), 7.19 (2H, d, J = 4.2 Hz), 7.11–7.06 (2H, m), 6.97–6.94 (1H, m), 6.27–6.25 (1H, m), 6.18 (1H, d, J = 9.6 Hz), 6.07 (1H, d, J = 9.0 Hz), 5.63 (1H, d, J = 16.8 Hz), 5.23 (1H, d, J = 10.8 Hz), 3.15 (1H, d, J = 13.8 Hz), 3.06 (1H, d, J = 13.8 Hz), 2.08 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 198.4, 169.3, 141.2, 140.5, 138.3, 135.3, 135.2, 132.3, 131.3, 127.7, 127.1, 125.9, 123.0, 115.8, 81.3, 40.1, 20.5; IR (CHCl3, cm−1): 2927, 2856, 1739, 1676; HRMS (DART+): m/z [M + H]+ Calcd for C17H17O3: 269.11777. Found: 269.11855.
The IMDA Reaction of 7e to 9e (Table 1, Entry 5)A solution of 7e (59 mg, 0.22 mmol) in xylenes (1 mL) was refluxed for 16 h. After cooling to room temperature, the crude products were purified by column chromatography on silica gel (EtOAc/Hexane) to provide the product 9e (43 mg, 0.16 mmol, 52%). Compound 9e: mp: 97.5–98.5 °C (recryst. from CH2Cl2); 1H-NMR (CDCl3, 600 MHz) δ: 7.14–7.06 (4H, m), 6.66 (1H, t, J = 7.2 Hz), 6.21 (1H, t, J = 7.2 Hz), 4.28 (1H, d, J = 15.6), 3.86 (1H, t, J = 5.4 Hz), 3.56 (1H, t, J = 6.0 Hz), 3.15 (1H, d, J = 16.2 H), 2.99 (1H, dd, J = 10.2, 4.8 Hz), 2.29 (1H, dd, J = 13.2, 4.2 Hz), 2.12 (3H, s), 1.84 (1H, dd, J = 11.4, 12.6 Hz); 13C-NMR (CDCl3, 150 MHz) δ: 203.1, 170.4, 144.3, 139.7, 134.9, 130.4, 128.1, 126.8, 126.6, 126.0, 80.8, 57.0, 43.7, 38.9, 36.3, 32.4, 22.0; IR (CHCl3, cm−1): 3024, 1736; HRMS (DART+): m/z [M + H]+ Calcd for C17H17O3: 269.11777. Found: 269.11805.
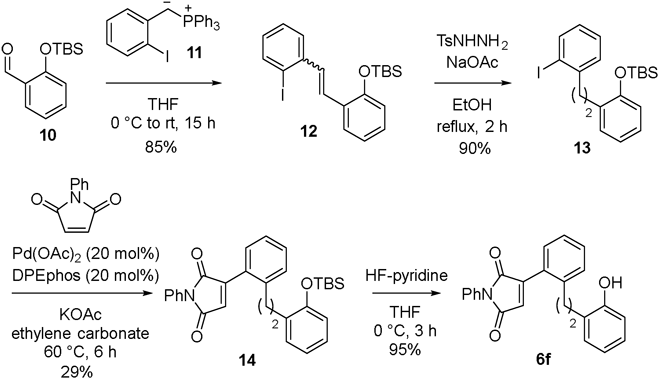
1-(tert-Butyldimethylsilyloxy)-2-(2-iodostyryl)benzene (12)To a cooled suspension of triphenyl(2-vinylbenzyl)phosphonium bromide (5.19 g, 9.28 mmol) in THF (46 mL) at 0 °C was added potassium tert-butoxide (1.21 g, 10.8 mmol) under argon. After 30 min, 2-tert-butyldimethylsiloxybenzaldehyde 10 (1.83 g, 7.73 mmol) was added over 10 min. The reaction temperature was warmed to r.t. and the mixture was stirred for 12 h. After adding water (30 mL) slowly, the aqueous phase was separated and extracted with EtOAc. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (Hexane) to give the product 12 (2.77 g, 6.34 mmol, 82%, a mixture of (E)- and (Z)-12) as a yellow oil. Compound 12: 1H-NMR (CDCl3, 600 MHz, E/Z = 5 : 1, data of (E)-12 is shown here) δ: 7.87 (1H, d, J = 9.0 Hz), 7.69 (1H, d, J = 7.2 Hz), 7.60 (1H, d, J = 7.8 Hz), 7.37 (1H, d, J = 10.8 Hz), 7.34 (1H, dd, J = 7.8, 7.8 Hz), 7.24 (1H, d, J = 16.2 Hz), 7.17 (1H, dd, J = 6.6, 6.6 Hz), 7.00 (1H, dd, J = 7.8, 7.8 Hz), 6.94 (1H, dd, J = 6.6, 6.6 Hz), 6.84 (1H, d, J = 8.4 Hz), 1.04 (9H, s), 0.24 (6H, s); 13C-NMR (CDCl3, 150 MHz, data of (E)-12 is shown here) δ: 153.3, 140.7, 139.6, 131.9, 128.9, 128.7, 128.4, 126.7, 126.4, 126.0, 121.6, 119.7, 100.5, 25.8, 18.3, −4.1; IR (neat, cm−1):1597, 1252, 1011; HRMS (DART+): m/z [M + H]+ Calcd for C20H26IOSi: 437.07976. Found: 437.08039.
1-(tert-Butyldimethylsilyloxy)-2-(2-iodophenethyl)benzene (13)Tosylhydrazide (50 mg, 0.12 mmol) and sodium acetate (47 mg, 0.57 mmol) were added to a solution of 12 (50 mg, 0.12 mmol) in EtOH (0.5 mL). The mixture was refluxed for 2 h with stirring. When the reaction was complete, the mixture was cooled to room temperature. The mixture was diluted with Et2O (5 mL) and washed with saturated solution of NaHCO3 and brine. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (EtOAc/hexane) to give 13 (45.6 mg, 0.104 mmol, 90%) as a yellow oil. Compound 13: 1H-NMR (CDCl3, 600 MHz) δ: 7.82 (1H, d, J = 7.8 Hz), 7.23 (1H, dd, J = 7.2, 7.2 Hz), 7.14 (1H, d, J = 6.0 Hz), 7.13 (1H, d, J = 7.8 Hz), 6.89–6.84 (2H, m), 6.81 (1H, d, J = 7.8 Hz), 2.98 (2H, dd, J = 10.8, 9.0 Hz), 2.87 (2H, dd, J = 10.8, 9.0 Hz), 1.03 (9H, s), 0.26 (6H, s); 13C-NMR (CDCl3, 150 MHz) δ: 153.6, 144.5, 139.4, 131.7, 130.4, 129.4, 128.4, 128.2, 127.7, 127.0, 120.1, 118.4, 100.8, 41.2, 30.9, 25.9, 18.3, −4.1; IR (neat, cm−1): 1252, 1011; HRMS (DART+): m/z [M + H]+ Calcd for C20H28IOSi: 439.09541. Found: 439.09442.
3-(2-(2-((tert-Butyldimethylsilyl)oxy)phenethyl)phenyl)-1-phenyl-1H-pyrrole-2,5-dione (14)A dry 30 mL flask containing a magnetic stirring bar was charged with 13 (900 mg, 2.05 mmol), Pd(OAc)2 (92 mg, 0.41 mmol), DPEphos (221 mg, 0.410 mmol), N-phenylmaleimide (1.78 g, 2.05 mmol), KOAc (604 mg, 6.15 mmol) and dry ethylene carbonate (5.4 g). The mixture was stirred at 60 °C for 6 h, and the mixture was cooled to room temperature. The crude product was purified by column chromatography on silica gel (EtOAc/Hexane) to give 14 (259 mg, 0.59 mmol, 29%) as a yellow oil. Compound 14: 1H-NMR (CDCl3, 600 MHz) δ: 7.48 (2H, dd, J = 9.0, 9.0 Hz), 7.41–7.28 (7H, m), 7.07–7.04 (1H, m), 6.82–6.75 (3H, m), 3.03 (2H, t, J = 7.2 Hz), 2.90 (2H, t, J = 7.2 Hz), 0.99 (9H, s), 0.21 (6H, s); 13C-NMR (CDCl3, 150 MHz) δ: 169.2, 169.1, 153.7, 145.7, 141.3, 131.7, 131.3, 130.6, 130.40, 130.35, 130.12, 130.09, 129.6, 129.0, 128.4, 128.2, 128.0, 127.6, 127.55, 127.46, 126.03, 125.91, 33.4, 32.6, 29.7, 25.8, 18.2, −4.1; IR (CHCl3, cm−1): 3473, 1768, 1714, 1623, 1598, 1502, 1257; HRMS (DART+): m/z [M + H]+ Calcd for C30H34NO3Si: 484.23079. Found: 484.23058.
3-(2-(2-Hydroxyphenethyl)phenyl)-1-phenyl-1H-pyrrole-2,5-dione (6f)To a solution of 14 (50 mg, 0.10 mmol) in THF (1 mL) was added pyridine (0.15 mL) and HF-pyridine (0.15 mL) at 0 °C. The solution was stirred at 0 °C for 3 h. The reaction mixture was diluted with EtOAc and H2O. The organic phase was washed with a saturated aqueous solution of NH4Cl, a saturated aqueous solution of NaHCO3, and brine. After the organic phase was concentrated under reduced pressure, the crude product was purified by column chromatography on silica gel (EtOAc/hexane) to yield 6f (35 mg, 0.095 mmol, 95%) as a yellow oil. Compound 6f: 1H-NMR (CDCl3, 600 MHz) δ: 7.49 (2H, dd, J = 9.0, 9.0 Hz), 7.45–7.37 (6H, m), 7.33–7.30 (1H, m), 7.07 (1H, dd, J = 9.0, 9.0 Hz), 7.00 (1H, dd, J = 7.2, 1.8 Hz), 6.82 (1H, dd, J = 6.6, 6.6 Hz), 6.69 (1H, s), 6.69 (1H, d, J = 6.6 Hz), 5.00 (1H, s), 3.00 (2H, dd, J = 10.2, 9.0 Hz), 2.90 (2H, dd, J = 10.8, 9.0 Hz); 13C-NMR (CDCl3, 150 MHz) δ: 169.7, 169.6, 153.7, 145.5, 141.3, 131.6, 130.7, 130.45, 130.41, 130.3, 129.1, 128.6, 128.0, 127.8, 127.7, 127.1, 126.2, 126.0, 120.8, 115.3, 34.2, 33.0; IR (CHCl3, cm−1): 3588, 3473, 3286, 1770, 1712, 1596, 1502; HRMS (DART+): m/z [M + H]+ Calcd for C24H20NO3: 370.14432. Found: 370.14448.
The Wessely Oxidation of 6f to 7f (Chart 7)To a solution of 6f (98 mg, 0.26 mmol) in AcOH (4 mL) was added Pb(OAc)4 (235 mg, 0.529 mmol) at 0 °C, and the reaction temperature was allowed to reach room temperature. After 30 min, the precipitate was filtered off with EtOAc, and the filtrate was washed with saturated aqueous solution of NaHCO3. The organic phase was dried over anhydrous MgSO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/Hexane) to yield 7f (39 mg, 0.090 mmol, 34%) as a yellow oil. Compound 7f: 1H-NMR (CDCl3, 600 MHz) δ: 7.52–7.50 (2H, m), 7.47–7.45 (2H, m), 7.41–7.39 (3H, m), 7.33–7.29 (2H, m), 6.77 (1H, s), 6.31–6.26 (2H, m), 6.15 (1H, d, J = 10.2 Hz), 2.82 (1H, ddd, J = 13.2, 13.2, 4.8 Hz), 2.73 (1H, ddd, J = 13.2, 13.2, 4.8 Hz), 2.14–2.07 (1H, m), 2.11 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 169.5, 169.4, 169.0, 145.6, 141.0, 140.5, 140.0, 130.6, 130.5, 130.0, 129.1, 128.6, 128.0, 127.9, 126.8, 126.6, 126.1, 123.0, 81.1, 39.0, 26.5, 20.4; IR (CHCl3, cm−1): 3473, 1770, 1737, 1716, 1675, 1502, 1390; HRMS (DART+): m/z [M + H]+ Calcd for C26H22NO5: 428.14980. Found: 428.15020.
The IMDA Reaction of 7f to 9f (Chart 7)Compound 7f (38 mg, 0.089 mmol) was heated at 170 °C in a vial without any solvent for 3.5 h. After cooling to room temperature, the crude products were purified by column chromatography on silica gel (EtOAc/Hexane) to provide the product 9f (20 mg, 0.047 mmol, 52%). Compound 9f: mp: 284.0–285.0 °C (recryst. from CH2Cl2); 1H-NMR (CDCl3, 600 MHz) δ: 8.61 (1H, d, J = 10.8 Hz), 7.49–7.47 (2H, m), 7.44–7.41 (1H, m), 7.34 (1H, dd, J = 7.2, 7.2 Hz), 7.27–7.20 (3H, m), 7.13 (1H, d, J = 7.8 Hz), 6.56 (1H, dd, J = 6.6, 6.6 Hz), 6.34 (1H, ddd, J = 7.8, 7.8, 1.2 Hz), 5.06 (1H, ddd, J = 5.1, 5.1, 1.8 Hz), 4.76 (1H, d, J = 4.2 Hz), 3.79 (1H, d, J = 6.0 Hz), 3.70–3.64 (1H, m), 3.17 (1H, ddd, J = 12.0, 6.6, 2.4 Hz), 2.92 (1H, ddd, J = 20.8, 6.6, 2.4 Hz), 2.73 (1H, ddd, J = 13.2, 13.2, 4.8 Hz), 2.36 (1H, ddd, J = 13.2, 13.2, 6.0 Hz), 2.00 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 201.0, 176.0, 174.0, 169.7, 136.5, 136.3, 134.6, 133.9, 131.5, 129.3, 129.1, 128.9, 128.5, 128.1, 126.5, 126.3, 79.6, 60.3, 51.4, 43.6, 39.2, 34.3, 33.9, 22.1,; IR (CHCl3, cm−1):3477, 1779, 1737, 1712, 1498, 1386; HRMS (DART+): m/z [M + H]+ Calcd for C26H22NO5: 428.14980. Found: 428.14995.
tert-Butyl(2-(2-iodo-3,4,5-trimethoxystyryl)phenoxy)dimethylsilane (16)To triphenylphosphine (2.1 g, 11 mmol) in a flask under a nitrogen atmosphere and heated to 100 °C in oil bath was added 1-(bromomethyl)-2-iodo-3,4,5-trimethoxybenzene (1.9 g, 4.9 mmol). After 10 min, to this was added CH2Cl2, and the mixture was concentrated, then filtered and washed extensively with EtOAc and CHCl3 to yield the phosphonium salt (2.8 g, 4.8 mmol, 97%) as a white solid. mp: 224.0–225.0 °C (recryst. from CH2Cl2); 1H-NMR (CDCl3, 600 MHz) δ: 7.56–7.89 (15H, m), 7.16 (1H, br s), 5.54–5.79 (2H, br m), 3.84 (3H, br d, J = 1.4 Hz), 3.72 (3H, br d, J = 1.4 Hz), 3.58 (3H, brd, J = 2.1 Hz); 13C-NMR (CDCl3, 150 MHz) δ: 142.2, 135.2, 134.5, 130.2, 125.6, 117.5, 117.0, 112.3, 92.8, 61.1, 60.6, 56.4, 35.6; IR (neat, cm−1):2939, 2854, 1482, 1483; HRMS (DART+): m/z [M-C10H12O3I]+ Calcd for C18H16P: 263.0990. Found: 263.0980.
To a cooled suspension of a phosphonium bromide (2.0 g, 3.1 mmol) in THF (15 mL) was added potassium tert-butoxide (404 mg, 3.60 mmol) under nitrogen at 0 °C. After stirring the mixture for 30 min, 2-tert-butyldimethylsiloxybenzaldehyde 10 (607 mg, 2.57 mmol) was added over 10 min. The reaction temperature was warmed to room temperature and the mixture was stirred for 12 h. After adding water (30 mL), the aqueous phase was separated and extracted with EtOAc. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (Hexane) to give 16 (1.29 g, 2.44 mmol, 95%, a mixture of (E)-16 and (Z)-16) as a colourless oil. Compound 16: 1H-NMR (CDCl3, 400 MHz) δ: 7.71 (0.4H, d, J = 8.0 Hz), 7.32 (0.8H, s), 7.17 (0.4H, t, J = 8.0 Hz), 6.96–7.11 (2.0H, m), 6.66–6.86 (2.2H, m), 6.51–6.60 (1.2H, m), 3.90 (5.4H, s), 3.87 (1.8H, s), 3.42 (1.8H, s), 1.06 (3.6H, s), 1.02 (5.4H, s), 0.25 (2.4H, s), 0.24 (s, 3.6H); 13C-NMR (CDCl3, 150 MHz) δ: 153.8, 153.6, 153.1, 153.0, 141.6, 141.0, 136.5, 133.1, 131.9, 130.5, 128.9, 128.5, 128.2, 127.9, 127.1, 126.1, 125.6, 121.6, 120.9, 119.5, 109.6, 104.9, 88.9, 87.8, 61.1, 61.0, 60.8, 60.7, 55.9, 55.6, 25.9, 25.8, 18.3, 4.08, −4.08, −4.22; IR (neat, cm−1): 2932, 2856, 1596, 1255; HRMS (DART+): m/z [M + H]+ Calcd for C23H32IO4Si: 527.1115. Found: 527.1106.
tert-Butyl(2-(2-iodo-3,4,5-trimethoxyphenethyl)phenoxy)dimethylsilane (17)Tosylhydrazide (1.04 g, 5.58 mmol) and sodium acetate (1.21 g, 14.0 mmol) were added to a solution of 16 (1.47 g, 2.79 mmol) in EtOH (13 mL), and the mixture was refluxed for 4 h. The reaction temperature was cooled to room temperature, and the mixture was diluted with Et2O. The mixture was washed with saturated aqueous solution of NaHCO3 and brine. The organic phase was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The products were purified by column chromatography on silica gel (EtOAc/Hexane) to give 17 (1.34 g, 2.54 mmol, 91%) as a colourless oil. Compound 17: 1H-NMR (CDCl3, 600 MHz) δ: 7.13–7.18 (1H, m), 7.06–7.11 (m, 1H), 6.85–6.90 (m, 1H), 6.79–6.83 (1H, m), 6.51 (1H, s), 3.88 (3H, s), 3.86 (3H, s), 3.74 (3H, s), 2.95–3.02 (2H, m), 2.82–2.89 (2H, m), 1.05 (9H, s), 0.27 (6H, s); 13C-NMR (CDCl3, 150 MHz) δ: 153.6, 153.3, 153.0, 140.3, 131.7, 130.6, 127.0, 121.1, 118.4, 108.8, 88.1, 61.0, 60.7, 55.9, 41.5, 31.0, 25.9, 18.3, −4.05; IR (CHCl3, cm−1): 2932, 2856, 1252; HRMS (DART+): m/z [M + H]+ Calcd for C23H34IO4Si: 529.1271. Found: 529.1270.
6-(2-((tert-Butyldimethylsilyl)oxy)phenethyl)-2,3,4-trimethoxybenzaldehyde (18)To a solution of 17 (500 mg, 0.95 mmol) in THF (10 mL) was added a solution of n-butyllithium (1.62 M in hexane, 1.46 mL, 2.37 mmol) dropwise at –78 °C. After stirring for 30 min at –78 °C, anhydrous DMF (0.73 mL, 9.5 mmol) was added dropwise. The reaction mixture was stirred for another 1 h at –78 °C. The reaction was warmed to room temperature and stirred for another 2 h, followed by the addition of 3M HCl (3 mL). The resulting mixture was extracted with Et2O, and the organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (EtOAc/Hexane) to afford compound 18 which was not completely pure. The crude product 18 was used without further purification in the next step.
(E)-tert-Butyldimethyl(2-(3,4,5-trimethoxy-2-(2-nitrovinyl)phenethyl)phenoxy)silane (19)The mixture of the crude 18, nitromethane (3.62 g, 59.4 mmol) and ammonium acetate (15 mg, 0.19 mmol) was stirred at 100 °C for 3 h. After cooling the reaction mixture to room temperature, the mixture was concentrated, and then H2O and dichloromethane were added. The mixture was washed with H2O, 1 N hydrochloric acid and brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (CH2Cl2/hexane) to yield 19 as a yellow solid (179 mg, 0.38 mmol, 40% yield for two steps). Compound 19: mp: 73.5–74.5 °C (recryst. from CH2Cl2); 1H-NMR (CDCl3, 600 MHz) δ: 8.11 (1H, d, J = 13.4 Hz), 7.92 (1H, d, J = 13.4 Hz), 7.07 (1H, t, J = 6.9 Hz), 6.97 (1H, d, J = 6.5 Hz), 6.83 (1H, t, J = 7.4 Hz), 6.78 (1H, d, J = 7.9 Hz), 6.45 (1H, s), 3.91 (3H, s), 3.85 (3H, s), 3.80 (3H, s), 3.03 (3H, t, J = 7.6 Hz), 2.86 (2H, t, J = 7.6 Hz), 1.03 (9H, s), 0.26 (6H, s); 13C-NMR (CDCl3, 150 MHz) δ: 156.0, 154.6, 153.6, 141.1, 140.6, 138.0, 132.2, 130.7, 130.6, 127.4, 121.2, 118.4, 115.3, 109.3, 60.9, 60.4, 55.8, 34.4, 32.9, 25.8, 18.2, −4.13 ; IR (CHCl3, cm−1): 2932, 2856, 1589, 1261; HRMS (DART+): m/z [M + H]+ Calcd for C24H36NO6Si: 474.23119. Found: 474.2310.
(E)-2-(3,4,5-Trimethoxy-2-(2-nitrovinyl)phenethyl)phenol (6g)To a solution of 19 (140 mg, 0.30 mmol) in THF (6 mL) was added TBAF (1.0 M solution in THF, 0.36 mL, 0.36 mmol) dropwise at 0 °C. The solution was stirred at 0 °C for 30 min. Water was added to the reaction mixture, and the resulting mixture was extracted with Et2O. The organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (EtOAc/Hexane) to yield 6g (104 mg, 0.029 mmol, 98%) as a yellow oil. Compound 6g: 1H-NMR (CDCl3, 600 MHz) δ: 8.27 (1H, d, J = 13.1 Hz), 7.94 (1H, d, J = 13.4 Hz), 7.05–7.10 (1H, m), 6.94 (1H, d, J = 7.6 Hz), 6.80 (1H, t, J = 7.4 Hz), 6.73 (1H, d, J = 7.9 Hz), 6.59 (1H, s), 5.18 (1H, brs), 3.90 (6H, s), 3.86 (3H, s), 3.06 (2H, t, J = 7.9 Hz), 2.86 (2H, t, J = 7.9 Hz); 13C-NMR (CDCl3, 150 MHz) δ: 156.2, 154.7, 153.7, 141.5, 140.7, 138.0, 132.9, 130.6, 127.7, 126.8, 120.8, 115.7, 115.1, 109.3, 60.9, 60.4, 56.0, 34.3, 33.7; IR (CHCl3, cm−1): 3444, 2938, 2850, 1589; HRMS (DART+): m/z [M + H]+ Calcd for C19H22NO6Si: 360.14471. Found: 360.14454.
The Wessely Oxidation of 6g to 7g (Table 2, Entry 1)Compound 6g (300 mg, 0.84 mmol) in AcOH (5 mL) was added to a solution of Pb(OAc)4 (556 mg, 1.25 mmol) in AcOH (5 mL) at 0 °C. The reaction temperature was warmed to room temperature, and the mixture was stirred for 2 h. The precipitate was filtered off with AcOEt and the filtrate was wash with saturated aqueous solution of NaHCO3. The organic phase was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (EtOAc/hexane) to yield 7g (151 mg, 0.36 mmol, 43%) as a yellow amorphous. Compound 7g: 1H-NMR (CDCl3, 600 MHz) δ: 8.11 (1H, d, J = 13.2 Hz), 7.98 (1H, d, J = 12.6 Hz), 7.04–7.09 (1H, m), 6.51 (1H, s), 6.44–6.48 (1H, m), 6.38–6.42 (1H, m), 6.24 (1H, d, J = 9.6 Hz), 3.94 (3H, s), 3.89 (3H, s), 3.84 (3H, s), 2.83–2.93 (1H, m), 2.74–2.82 (1H, m), 2.19 (3H, s), 1.99–2.01 (1H, m), 1.82–1.92 (1H, m); 13C-NMR (CDCl3, 150 MHz) δ: 198.1, 169.6, 156.4, 155.0, 140.9, 140.8, 139.8, 138.5, 131.8, 126.8, 123.5, 114.9, 109.2, 80.9, 60.9, 60.5, 56.1, 39.6, 27.5, 20.4; IR (CHCl3, cm−1): 2927, 2854, 1749, 1716, 1595; HRMS (DART+): m/z [M + H]+ Calcd for C21H24NO8: 418.1502. Found: 418.1507.
The IMDA Reaction of 7g to 9g (Table 2, Entry 3)A solution of 7g (39 mg, 0.092 mmol) in 2,2,2-trifluoroethanol (1 mL) was heated in a vial at 130 °C under microwave irradiation for 18 h. After concentration, the crude product was purified by column chromatography on silica gel (EtOAc/CH2Cl2) to provide 9g (9.6 mg, 0.023 mmol, 25%) as a brown solid. Compound 9g: mp: 84.0–85.5 °C (recryst. from CH2Cl2); 1H-NMR (CDCl3, 600 MHz) δ: 6.38 (1H, t, J = 7.2 Hz), 6.35 (1H, s), 6.31(1H, t, J = 6.9 Hz), 5.81 (1H, d, J = 3.6 Hz), 5.21 (1H, s), 4.89 (1H, d, J = 4.2 Hz), 3.97 (3H, s), 3.86 (3H, s), 3.83–3.89 (1H, m), 3.82 (3H, s), 3.39 (1H, t, J = 5.2 Hz), 3.23–3.35 (1H, m), 3.00–3.09 (1H, m), 2.71–2.81 (1H, m), 2.24–2.33 (1H, m), 2.02 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 202.3, 170.2, 152.8, 152.0, 141.8, 133.1, 131.9, 129.0, 121.8, 112.3, 82.9, 79.9, 61.8, 61.1, 56.1, 53.7, 42.5, 36.1, 33.3, 33.0, 22.5; IR (CHCl3, cm−1):2940, 2838, 1736, 1552; HRMS (DART+): m/z [M + H]+ Calcd for C21H24NO8: 418.1502. Found: 418.1505.
Computation DetailsGeometry optimization was performed with Spartan’1837) and the Gaussian 09 packages.38) The ground-state geometries of all compounds were determined by means of the following successive steps: Conformational search with MMFF94,39) then DFT calculation with B3LYP functionals.40) The transition-state geometries of all compounds were determined by the DFT calculation with B3LYP functionals followed by IRC calculations.41,42) The basis set employed for DFT geometry optimization was the native 6–31G(d, p) for Fig. 1(a) or the native 6–31G(d) for Fig. 1(b)–(d).