Regular Articles
Development of Nitrolactonization Mediated by Iron(III) Nitrate Nonahydrate
2020 Volume 68 Issue 12 Pages 1220-1225
Details
2020 Volume 68 Issue 12 Pages 1220-1225
The nitrolactonization of alkenyl carboxylic acids mediated by Fe(NO3)3·9H2O has been developed. Nitrolactones were obtained in up to 93% yield by treatment of alkenyl carboxylic acids with Fe(NO3)3·9H2O. Mechanistic studies disclosed that the reaction proceeded through a radical intermediate generated from addition of NO2 to alkenyl carboxylic acids.
Nitro compounds are useful synthons in synthetic organic chemistry due to their availability for various transformations. The strong electron-withdrawing property of the nitro group plays an important role in C–C bond formation reactions1,2) including the nitro–aldol reaction (Henry reaction),3–5) Michael reaction,6–11) and alkylation.12,13) Nitrones and nitrile oxides generated from nitro compounds undergo 1,3-dipole cycloaddition reaction to give various heterocycles.14–16) The nitro group can be converted into a carbonyl or amino group through the Nef reaction17,18) or reduction, respectively.19) For these reasons, nitro compounds are heavily used as versatile starting materials in the synthesis of natural products and drugs.20–25) Nitration mediated by iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O) has been recently reported.26–35) The use of Fe(NO3)3·9H2O has attracted attention as a source of nitrogen dioxide in organic synthesis due to its properties as a readily available reagent of low toxicity and cost. It has been suggested that these nitrations proceed via a radical intermediate derived by addition of nitrogen dioxide generated from Fe(NO3)3·9H2O with heating.36) We envisioned the novel development of a nitrolactonization of styrene derivative 1 mediated by Fe(NO3)3·9H2O (Chart 1). Because nitrolactone 2 could lead to 1,2-aminoalcohol and amino acid derivatives bearing a tetrasubstituted carbon center, the development of a new nitrolactonization reaction leading to substituted nitrolactones would be useful not only in natural product synthesis but in medicinal chemistry.
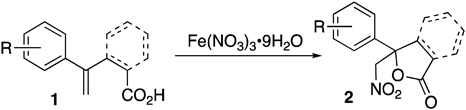
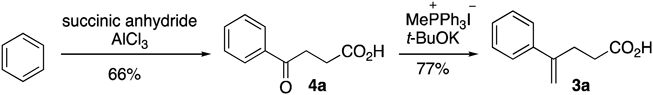
Precursor 3a for nitrolactonization was prepared from benzene by Friedel–Crafts acylation with succinic anhydride followed by a Wittig reaction of the resultant ketone 4a (Chart 2).
Nitrolactonization of carboxylic acid 3a was investigated according to the reported method of generating NO2 from Fe(NO3)3·9H2O37) (Table 1). Treatment of 3a with 0.5 equivalent (equiv.) of Fe(NO3)3·9H2O in 1,2-dichlorobenzene at 130 °C for 4 h afforded nitrolactone 5a, hydroxylactone 6a, and lactone 7a in 48, 3, and 7% yields, respectively (entry 1). The nitration regioselectively took place at the olefinic moiety of 3a to produce 5a, with the product structure confirmed by X-ray crystallographic analysis.38) Hydroxylactone 6a was envisioned to be produced via a nitrite lactone intermediate due to the ambident property of NO2 while lactone 7a was likely produced via proto-lactonization mediated by HNO3 that was generated from NO2 gas and water.39) The effect of the metal nitrate was investigated by use of copper(II) nitrate, zinc nitrate, and silver nitrate in the reaction (entries 2–4). These metal nitrates afforded nitrolactone 5 in lower yield when compared to the reaction with Fe(NO3)3 (entry 1 vs. entries 2–4). Reducing or increasing the number of equivalents of Fe(NO3)3 resulted in diminished yields (entry 1 vs. entries 5 and 6). The differences in the yields of products involved in the number of equivalent of Fe(NO3)3 would attribute the concentration of NO2 generated for Fe(NO3)3. Lower equivalent of Fe(NO3)3 would not be enough to undergo NO2 addition reaction but lead to decomposition of 5a probably due to the equilibrium mixture between NO2 and its various reactive isomers of N2O4 (entry 5).40–43) Higher equivalent of Fe(NO3)3 would generate a high concentration of HNO3, which lead to proto-lactonization rather than radical addition of NO2 (entry 6). To investigate the assumption that the heating-up time and reaction temperature would affect the reaction progress and yield, the reaction mixture was quickly heated (entries 7–9). When the reaction was conducted in a pre-heated oil bath at 130 °C, nitrolactone 5a was produced in an improved yield of 58% and the reaction time was shortened (entry 1 vs. entry 7). Addition of KHCO3 as a base to suppress the formation of byproduct 7a did not result in an improved yield of 5a (entry 8). Addition of proton-sponge® resulted in a complex product mixture (entry 9).
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Metal nitrate | X | Time (h) | Yield (%) | ||
| 5a | 6a | 7a | ||||
| 1 | Fe(NO3)3·9H2O | 0.5 | 4 | 48 | 3 | 7 |
| 2 | Cu(NO3)2·3H2O | 0.75 | 5 | 36 | 12 | 4 |
| 3 | Zn(NO3)2·6H2O | 0.75 | 3 | 8 | 8 | 30 |
| 4 | AgNO3 | 1.5 | 3 | 3 | 4 | 0 |
| 5 | Fe(NO3)3·9H2O | 0.34 | 4 | 7 | 4 | 5 |
| 6 | Fe(NO3)3·9H2O | 0.67 | 4.5 | 24 | 3 | 49 |
| 7b) | Fe(NO3)3·9H2O | 0.5 | 1.5 | 58 | 5 | 13 |
| 8b,c) | Fe(NO3)3·9H2O | 0.5 | 2.5 | 54 | approx. 10d) | 7 |
| 9b,e) | Fe(NO3)3·9H2O | 0.5 | 24 | 0 | 0 | 0 |
a) A stirring mixture of metal nitrate and 3 in 1,2-dichlorobenzen was heated gradually from room temperature (r.t.) to 130 °C and after temperature reached at 130 °C, the reaction was run for the indicated time. b) The reaction was performed in pre-heated oil bath (130 °C). c) 0.3 equiv. of KHCO3 was added. d) A small amount of 4-phenyl-4-oxobutanoic acid was contained. e) 1.0 equiv. of proton-sponge® was added.
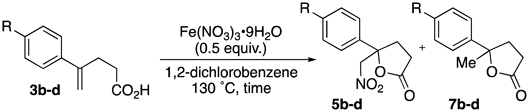
The effect of substitution at the phenyl group of starting material 3 was examined (Table 2). Treatment of 3b with Fe(NO3)3·9H2O in 1,2-dichlorobenzene at 130 °C afforded nitrolactone 5b and lactone 7b in 12 and 69% yields, respectively, without production of hydroxylactone. The yield of 7b was higher than that of 5b because the electron-donating effect of the methoxy group prompted a protonation reaction instead of the radical addition of nitrogen dioxide (entry 1). Substituents bearing electron-withdrawing groups such as NO2 and CN at phenyl group resulted in low yields of 5c and 5d due to complication in reaction (entries 2 and 3).
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Starting material | R | Time (h) | Products | Yield (%) | |
| 5 | 7 | |||||
| 1 | 3b | OMe | 3 | 5b, 7b | 12 | 69 |
| 2 | 3c | NO2 | 2 | 5c | 16 | 0 |
| 3 | 3d | CN | 4 | 5d | 26 | 0 |
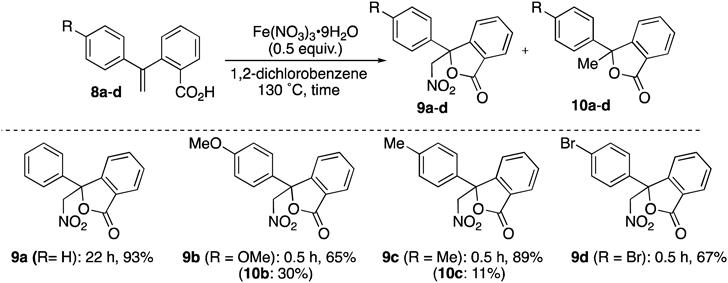
The nitrolactonization of benzoic acid derivatives 8a–d was investigated to determine whether yields would improve due to acceleration of the cyclization rate and stabilization of the radical intermediate (vide infra) (Table 3). The reaction of 8a for 22 h proceeded smoothly to give 9a44) as a sole product in 93% yield. Nitrolactonization of compounds 8b and 8c bearing electron-donating substituents at the aromatic ring produced not only 9b (65% yield) and 9c (89% yield), respectively, but also 10b (30% yield) and 10c (11% yield), respectively, as a result of addition of nitrogen dioxide and the competing formation of a stable cation at the double benzylic position. Substitution of a bromo group at the aromatic ring, which can be converted into various functional groups, resulted in a reduced yield of nitrolactone 9d (67%) but lactone 10d was not observed.
 |
A plausible mechanism is shown in Chart 3. Nitrogen dioxide generated from Fe(NO3)3·9H2O by pyrolysis would react with 3a at nitrogen atom to lead to radical intermediate A. Oxidation of A, which might be mediated by Fe(NO3)3 and/or HNO3, would give carbocation B, which would undergo cyclization to form 5a. This radical mechanism is supported by the fact that nitrolactonization of 8a in the presence of 2,6-di-tert-butyl-4-methylphenol (BHT) as a radical scavenger reduced the yield of 9a (12%) (Table 3, entry 1 vs. Chart 4) and recovered 8a in 16% yield. Isolation of 8a and 9a was possible although other products could be unidentified because of complication in the reaction. Through a similar process leading to production of 5a, hydroxylactone 6a would be obtained through nitrite radical intermediates C, carbocation D, and nitrite E, sequentially. It was assumed that cleavage of the O–NO bond of E took place in the reaction media or by addition of water during the workup stage. The ambident property of NO2 might produce nitrite intermediate C. It had been reported not only that partial pressure of oxygen influenced the production of nitrite adducts in the reaction of isobutylene and N2O4 which has been regarded as an equilibrium mixture with NO2,40–43,45) but also that oxidative cleavage of a tetrasubstituted olefin catalyzed by Fe(NO3)3 would proceed via a dinitrite intermediate in the presence of molecular oxygen.37) We speculated that production of 6a might be related to the amount of dissolved oxygen in the reaction media, however the detailed effect of the dissolved oxygen on the production of 6a was unclear. Lactone 7a was obtained via carbocation F by catalysis of nitric acid generated from H2O and NO2 in the reaction media.
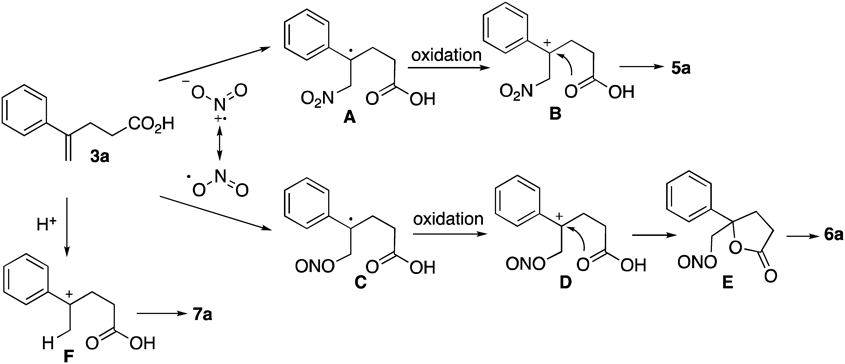

The nitrolactonization of carboxylic acids mediated by Fe(NO3)3·9H2O was developed. Mechanistic studies disclosed that the reaction proceeded via a radical intermediate derived from addition of NO2, which was generated by pyrolysis of Fe(NO3)3·9H2O, at the olefin moiety. These results demonstrate the utility of Fe(NO3)3·9H2O as a source of nitrogen dioxide and could lead to the development of new reactions mediated by Fe(NO3)3·9H2O.
1H-NMR were measured in CDCl3 or dimethyl sulfoxide (DMSO)-d6 solution and referenced from tetramethylsilane (TMS) (0.00 ppm) using JEOL JNM ECA-600 (600 MHz) or JEOL JNM ECA-400 (400 MHz) spectrophotometer, unless otherwise noted. 13C-NMR were measured in CDCl3 solution and referenced to CDCl3 (77.0 ppm), DMSO-d6 ((CD3)3SO) (39.5 ppm) or CD3OD (49.0 ppm) using JEOL JNM ECA-600 (150 MHz) or JEOL JNM ECX-400 (100 MHz) spectrophotometer, unless otherwise noted. Chemical shifts are reported in ppm. When peak multiplicities are reported, the following abbreviations are used: s, singlet; d, doublet; t, triplet, q, quartet; m, multiplet; br, broadened. IR spectra were recorded on Horiba IR-710 spectrometer. Mass spectra were obtained on JEOL JMS-T100TD. Flash column chromatography was performed on Silica Gel (SiliaFlash® F60). TLC was performed on precoated plates (0.25 mm, silica gel Merck Kieselgel 60F245), and compounds were visualized with UV light and p-anisaldehyde stain or phosphomolybdic acid stain. Melting points were measured with Yanaco MICRO MELTING POINTAPPARATUS. 1,2-Dichlorobenzene was purchased from TOKYO CHMICAL INDUSTRY CO., LTD. (Japan) and used without any purifications. Fe(NO3)3·9H2O was purchased from FUJIFILM Wako Pure Chemical Corporation (Japan) and used without any purifications.
Compounds 3a,b46)3c,47)3d,48) and, 8a–d49) were prepared according to the literatures and identified with 1H-NMR by comparison with the reported ones.
General Procedure 1: Optimization of Reaction Conditions (Table 1)A mixture of 3a (47.0 mg, 0.27 mmol) and metal nitrate in 1,2-dichlorobenzene (2.7 mL) was stirred at 130 °C. After being stirred for the indicated time in Table 1, the reaction was quenched by addition of sat. aq. NaHCO3 and the resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 8/2 to 7/3) to give 5a, 6a, and 7a, respectively.
Entry 1According to the general procedure 1, the reaction was performed with Fe(NO3)3·9H2O (54.0 mg, 0.13 mmol) for 4 h to give 5a (28.6 mg, 48%), 6a (1.7 mg, 3%), and 7a (3.5 mg, 7%), respectively.
Entry 2According to the general procedure 1, the reaction was performed with Cu(NO3)2·3H2O (48.3 mg, 0.2 mmol) for 5 h to give 5a (21.0 mg, 36%), 6a (6.4 mg, 12%), and 7a (1.7 mg, 4%), respectively.
Entry 3According to the general procedure 1, the reaction was performed with Zn(NO3)2·6H2O (59.5 mg, 0.2 mmol) for 3 h to give 5a (4.6 mg, 8%), 6a (3.9 mg, 8%), and 7a (14.2 mg, 30%), respectively.
Entry 4According to the general procedure 1, the reaction was performed with AgNO3 (68.0 mg, 0.4 mmol) for 3 h to give 5a (1.6 mg, 3%), 6a (1.9 mg, 4%), and recovered 3a (1.2 mg, 3%), respectively.
Entry 5According to the general procedure 1, the reaction was performed with Fe(NO3)3·9H2O (36.7 mg, 0.09 mmol) for 4 h to give 5a (4.3 mg, 7%), 6a (2.1 mg, 4%), and 7a (2.3 mg, 5%), respectively.
Entry 6According to the general procedure 1, the reaction was performed with Fe(NO3)3·9H2O (72.0 mg, 0.18 mmol) for 4.5 h to give 5a (14.4 mg, 24%), 6a (1.5 mg, 3%), and 7a (23.2 mg, 49%), respectively.
Entry 7A mixture of 3a (44.0 mg, 0.25 mmol) and Fe(NO3)3·9H2O (50.5 mg, 0.13 mmol) in 1,2-dichlorobenzene (2.5 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 1.5 h at same temperature, the mixture was cooled to r.t. and quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 9/1 to 3/2) to give 5a (31.8 mg, 58%), 6a (2.6 mg, 5%), and 7a (5.6 mg, 13%), respectively.
Entry 8A mixture of 3a (44.0 mg, 0.25 mmol), Fe(NO3)3·9H2O (50.5 mg, 0.13 mmol), and KHCO3 (7.5 mg, 0.075 mmol) in 1,2-dichlorobenzene (2.5 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 2.5 h at same temperature, the mixture was cooled to r.t. and quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 9/1 to 3/2) to give 5a (29.6 mg, 54%), 6a (4.8 mg, a small amount of 4-phenyl-4-oxobutanoic acid was contaminated, approx. 10%), and 7a (3.2 mg, 7%), respectively.
5-Nitromethyl-5-phenyloxolan-2-one (5a)White solid; mp: 95.0–95.5 °C; IR (CHCl3) cm−1: 1790, 1560; 1H-NMR (CDCl3, 400 MHz) δ: 7.47–7.38 (5H, m), 4.81 (1H, d, J = 12.6 Hz), 4.75 (1H, d, J = 12.6 Hz), 3.00–2.91 (1H, m), 2.75–2.66 (2H, m), 2.58–2.49 (1H, m); 13C-NMR (CDCl3, 150 MHz) δ: 174.7, 138.7, 129.1, 124.6, 84.6, 81.9, 31.8, 27.8; 13C-NMR (CD3OD, 150 MHz) δ: 177.5, 141.0, 129.9, 129.7, 125.9, 86.6, 82.9, 33.6, 28.5; high resolution (HR)MS (DART) m/z Calcd for C11H12NO4 [(M + H)+]: 222.07663. Found 222.07629.
5-Hydroxymethyl-5-phenyloxolan-2-one (6a)50)White solid; mp: 108.5–109.5 °C; IR (CHCl3) cm−1: 3953, 1716, 1686; 1H-NMR (CDCl3, 400 MHz) δ: 7.40–7.34 (5H, m), 3.88 (1H, dd, J = 5.2, 12.4 Hz), 3.73 (1H, dd, J = 8.4, 12.4 Hz), 2.84–2.70 (2H, m), 2.57–2.49 (1H, m), 2.43–2.39 (1H, m), 1.91 (1H, dd, J = 5.2, 8.4 Hz); MS (DART) m/z: 193 [(M + H)+]. The spectral data were identified with the reported ones.
5-Methyl-5-phenyloxolan-3-one (7a)51)Yellow oil; IR (CHCl3) cm−1: 1764, 1683; 1H-NMR (CDCl3, 400 MHz) δ: 7.38–7.29 (5H, m), 2.64–2.41 (4H, m), 1.73 (3H, s); MS (DART) m/z: 177 [(M + H)+]. The spectral data were identified with the reported ones.
Nitrolactonization of 3bA mixture of 3b (51.6 mg, 0.25 mmol) and Fe(NO3)3·9H2O (50.5 mg, 0.13 mmol) in 1,2-dichlorobenzene (2.5 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 3 h at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 8/1 to 6/4) to give 5b (7.8 mg, 12%) and 7b (35.7 mg, 69%), respectively.
5-(4′-Methoxyphenyl)-5-nitromethyloxolan-2-one (5b)Pale yellow solid; mp: 130.0–131.0 °C; IR (CHCl3) cm−1: 1790, 1558; 1H-NMR (CDCl3, 400 MHz) δ: 7.33 (2H, d, J = 9.0 Hz), 6.94 (2H, d, J = 9.0 Hz), 4.78 (1H, d, J = 12.4 Hz), 4.71 (1H, d, J = 12.4 Hz), 3.83 (3H, s), 2.92–2.87 (1H, m), 2.73–2.64 (2H, m), 2.58–2.51 (1H, m); HRMS (DART) m/z Calcd for C12H17N2O5 [(M + NH4)+]: 269.11375. Found 269.11193.
5-(4′-Methoxyphenyl)-5-methyloxolan-2-one (7b)52)Pale yellow oil; IR (CHCl3) cm−1: 1770, 1513, 1249; 1H-NMR (CDCl3, 400 MHz) δ: 7.30 (2H, d, J = 8.8 Hz), 6.90 (2H, d, J = 8.8 Hz), 3.81 (3H, s), 2.63–2.37 (4H, m), 1.70 (3H, s). The spectral data were identified with the reported ones.
Nitrolactonization of 3cA mixture of 3c (52.0 mg, 0.24 mmol) and Fe(NO3)3·9H2O (48.5 mg, 0.12 mmol) in 1,2-dichlorobenzene (2.4 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 2 h at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 5/1 to 6/4) to give 5-(4′-nitrophenyl)-5-nitromethyloxolan-2-one (5c) (10.1 mg, 16%) as pale yellow needles; mp: 141.0–142.0 °C (recrystallized from AcOEt/hexane); IR (KBr) cm−1: 1795, 1731, 1606, 1556, 1378; 1H-NMR (DMSO-d6, 600 MHz) δ: 8.29 (2H, d, J = 9.0 Hz), 7.75 (2H, d, J = 9.0 Hz), 5.52 (1H, d, J = 14.4 Hz), 5.39 (1H, d, J = 14.4 Hz), 2.80 (1H, ddd, J = 6.6, 9.6, 17.4 Hz), 2.71 (1H, ddd, J = 6.6, 9.6, 13.8 Hz), 2.55 (1H, ddd, J = 6.0, 10.2, 17.4 Hz), 2.39–2.35 (1H, m); 13C-NMR (DMSO-d6, 150 MHz) δ: 175.3, 147.6, 147.3, 126.3, 123.7, 84.3, 81.4, 32.9, 27.2; HRMS (DART) m/z Calcd for C11H11N2O6 [(M + H)+]: 267.06171. Found 267.06153.
Nitrolactonization of 3dA mixture of 3d (41.5 mg, 0.21 mmol) and Fe(NO3)3·9H2O (41.6 mg, 0.11 mmol) in 1,2-dichlorobenzene (2.1 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 4 h at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 6/4 to 1/1) to give 5-(4′-cyanophenyl)-5-nitromethyloxolan-2-one (5d) (13.3 mg, 26%) as pale yellow solid; mp: 174.5–175.0 °C; IR (neat) cm−1: 2231, 1788, 1631, 1612, 1556; 1H-NMR (methanol-d4, 400 MHz) δ: 7.81 (2H, d, J = 8.8 Hz), 7.66 (2H, d, J = 8.8 Hz), 5.20 (1H, d, J = 13.6 Hz), 5.14 (1H, d, J = 13.6 Hz), 2.88–2.70 (2H, m), 2.61–2.51 (2H, m); 13C-NMR (DMSO-d6, 150 MHz) δ: 175.4, 145.6, 132.6, 125.8, 118.4, 111.1, 84.3, 81.3, 32.8, 27.1; HRMS (DART) m/z Calcd for C12H14N3O4 [(M + NH4)+]: 264.09843. Found 264.09788.
Nitrolactonization of 8aA mixture of 8a (56.0 mg, 0.25 mmol) and Fe(NO3)3·9H2O (50.5 mg, 0.13 mmol) in 1,2-dichlorobenzene (2.5 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 22 h at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 9/1 to 3/1) to give 3-(nitromethyl)-3-phenylisobenzofuran-1(3H)-one (9a)44) (62.8 mg, 93%) as a white solid; mp: 128.5–130.0 °C; IR (CHCl3) cm−1: 1782, 1562; 1H-NMR (CDCl3, 400 MHz) δ: 7.96–7.40 (9H, m), 5.26 (1H, d, J = 12.8 Hz), 5.17 (1H, d, J = 12.8 Hz); 13C-NMR (CDCl3, 150 MHz) δ: 168.1, 147.5, 136.0, 134.7, 130.5, 129.6, 129.3, 126.5, 125.7, 125.1, 122.9, 85.5, 80.2; HRMS (DART) m/z Calcd for C15H12NO4 [(M + H)+]: 270.07663. Found 270.07407.
Nitrolactonization of 8bA mixture of 8b (35.6 mg, 0.14 mmol) and Fe(NO3)3·9H2O (28.3 mg, 0.07 mmol) in 1,2-dichlorobenzene (1.4 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 30 min at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 9/1 to 7/3) to give 3-nitromethyl-3-(4′-methoxypheyl)isobenzofuran-1(3H)-one (9b) (26.9 mg, 65%) and 3-methyl-3-(4′-methoxyphenyl)isobenzofuran-1(3H)-one (10b) (10.5 mg, 30%), respectively.
3-Nitromethyl-3-(4′-methoxyphenyl)isobenzofuran-1(3H)-one (9b)White solid; mp: 49.0–50.0 °C; IR (CHCl3) cm−1: 1780, 1560; 1H-NMR (CDCl3, 400 MH) δ: 7.96–7.60 (4H, m), 7.45 (2H, d, J = 9.2 Hz), 6.92 (2H, d, J = 9.2 Hz), 5.23 (1H, d, J = 13.0 Hz), 5.14 (1H, d, J = 13.0 Hz), 3.80 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 168.2, 160.3, 147.6, 134.6, 130.9, 127.7, 126.7, 126.3, 125.7, 123.0, 114.5, 85.5, 80.2, 55.3; HRMS (DART) m/z Calcd for C16H14NO5 [(M + H)+]: 300.08720. Found 300.08665.
3-Methyl-3-(4′-methoxyphenyl)isobenzofuran-1(3H)-one (10b)49)Pale yellow oil; IR (CHCl3) cm−1: 1759, 1514, 1255; 1H-NMR (CDCl3, 400 MHz) δ: 7.91 (1H, d, J = 8.0 Hz), 7.66 (1H, t, J = 7.6 Hz), 7.52 (1H, t, J = 7.6 Hz), 7.42 (1H, d, J = 8.0 Hz), 7.33 (2H, d, J = 9.0 Hz), 6.86 (2H, d, J = 9.0 Hz), 3.79 (3H, s), 2.03 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 170.0, 159.5, 154.4, 134.2, 132.5, 129.0, 126.6, 125.8, 125.2, 122.0, 113.9, 87.6, 55.3, 27.1; HRMS (DART) m/z Calcd for C16H15O3 [(M + H)+]: 255.10212. Found 255.10299. The spectral data were identified with the reported ones.
Nitrolactonization of 8cA mixture of 8c (59.6 mg, 0.25 mmol) and Fe(NO3)3·9H2O (50.5 mg, 0.13 mmol) in 1,2-dichlorobenzene (2.5 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 30 min at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The mixture was filtered through a pad of Celite and the filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residue was purified through silica gel column chromatography (hexane/AcOEt = 9/1 to 7/3) to give 3-nitromethyl-3-(p-tolyl)isobenzofuran-1(3H)-one (9c) (63.0 mg, 89%) and 3-methyl-3-(p-tolyl)isobenzofuran-1(3H)-one (10c) (7.0 mg, 11%), respectively.
3-Nitromethyl-3-(p-tolyl)isobenzofuran-1(3H)-one (9c)White solid; mp: 99.0–100.5 °C; IR (CHCl3) cm−1: 1780, 1560; 1H-NMR (CDCl3, 400 MHz) δ: 7.94 (1H, d, J = 7.6 Hz), 7.78–7.74 (1H, m), 7.72 (1H, d, J = 7.6 Hz), 7.63–7.59 (1H, m), 7.43 (2H, d, J = 8.2 Hz), 7.23 (2H, d, J = 8.2 Hz), 5.24 (1H, d, J = 13.0 Hz), 5.16 (1H, d, J = 13.0 Hz), 2.35 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 168.2, 147.7, 139.6, 134.6, 133.0, 130.3, 129.9, 126.3, 125.6, 124.9, 122.9, 85.5, 80.1, 21.0; HRMS (DART) m/z Calcd for C16H14NO4 [(M + H)+]: 284.09228. Found 284.09152.
3-Methyl-3-(p-tolyl)isobenzofuran-1(3H)-one (10c)Pale yellow oil; IR (CHCl3) cm−1: 1759, 1514; 1H-NMR (CDCl3, 400 MHz) δ: 7.90 (1H, d, J = 7.6 Hz), 7.67–7.63 (1H, m), 7.53–7.49 (1H, m), 7.44 (1H, d, J = 7.6 Hz), 7.32 (2H, d, J = 7.8 Hz), 7.16 (2H, d, J = 7.8 Hz), 2.33 (3H, s), 2.03 (3H, s); 13C-NMR (CDCl3, 150 MHz) δ: 170.0, 154.4, 138.2, 137.7, 134.2, 129.3, 129.0, 125.8, 125.08, 125.06, 122.0, 87.6, 27.2, 21.0; HRMS (DART) m/z Calcd for C16H15O2 [(M + H)+]: 239.10720. Found 239.10637.
Nitrolactonization of 8dA mixture of 8d (33.2 mg, 0.11 mmol) and Fe(NO3)3·9H2O (22.1 mg, 0.06 mmol) in 1,2-dichlorobenzene (1.1 mL) was lowered into pre-heated oil bath (130 °C). After being stirred for 30 min at same temperature, the mixture was cooled into r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite and the filtrate was extracted with CH2Cl2. The extracts were washed with brine, and dried over Na2SO4, filtered, and concentrated. The residual oil was purified through silica gel column chromatography (hexane/AcOEt = 9/1 to 7/3) to give 3-(4′-bromophenyl)-3-nitromethylisobenzofuran-1(3H)-one (9d) as white solid; mp: 104.0–104.5 °C; IR (CHCl3) cm−1: 1784, 1558; 1H-NMR (CDCl3, 400 MHz) δ: 7.96 (1H, d, J = 7.6 Hz), 7.80–7.76 (1H, m), 7.70 (1H, d, J = 7.6 Hz), 7.66–7.62 (1H, m), 7.57 (2H, d, J = 8.6 Hz), 7.45 (2H, d, J = 8.6 Hz), 5.22 (1H, d, J = 13.2 Hz), 5.12 (1H, d, J = 13.2 Hz); 13C-NMR (CDCl3, 150 MHz) δ: 167.8, 147.1, 135.1, 134.9, 132.5, 130.7, 126.8, 126.7, 125.5, 124.0, 122.7, 84.9, 80.0; HRMS (DART) m/z Calcd for C15H14BrN2O4 [(M + NH4)+]: 365.01369. Found 365.01524.
Experiment for Chart 4Di-tert-butylhydroxytoluene (BHT) (132.2 mg, 0.6 mmol) was added to a mixture of 8a (44.9 mg, 0.2 mmol) and Fe(NO3)3·9H2O (40.4 mg, 0.1 mmol) in 1,2-dichlorobenzene (2.0 mL). The mixture was lowered into pre-heated oil bath (130 °C). After being stirred for 19 h at same temperature, the mixture was cooled to r.t. and the reaction was quenched by addition of sat. aq. NaHCO3. The resulting mixture was filtered through a pad of Celite. The filtrate was acidified the 4N HCl and extracted with CH2Cl2. The extracts were washed with water, brine, and dried over Na2SO4, filtered, and concentrated. The residue was chromatographed on silica gel (hexane/AcOEt = 9/1 to 8/2) to give a mixture of 9a (12%) and 8a (16%), respectively. The yields of 9a and 8a were determined by qNMR using 1,4-dinitrobenzene as an internal standard.
This research was supported by JSPS KAKENHI Grant Number JP17K08208.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.