Abstract
Proteins incorporating artificial moieties such as fluorophores and drugs have enjoyed increasing use in chemical biology and drug development research. Preparation of such artificial protein derivatives has relied mainly on native chemical ligation in which peptide/protein thioesters chemoselectively react with N-terminal cysteine (Cys) peptides to afford protein molecules. The protein thioesters derived from expressed proteins represent thioesters that are very useful for the preparation of artificial proteins by native chemical ligation with synthetic peptides with N-terminal Cys. We recently have developed a traceless thioester-producing protocol using carboxypeptidase Y (CPaseY) which is compatible with an expressed protein. The traceless character is ensured by CPaseY-mediated hydrazinolysis of C-terminal Xaa (X)-Cys-proline (Pro)-leucine (Leu)-OH sequence followed by an auto-processing of the Cys-Pro (CP) dipeptide unit, affording the corresponding X-thioester (X-SR). However, hydrazinolysis of the amide bond in the prolyl leucine junction depends significantly on the nature of X. In the case of hydrophobic X residues, the hydrazinolysis overreacts to give several hydrazides while the reaction of hydrophilic X residues proceeds slowly. In this research, we attempted to develop an X-independent CPaseY-mediated protocol and found that the incorporation of a triple CP sequence into the C-terminal end (X-(CP)3-Leu-OH) allows for efficient X-SR formation in a manner that is independent of X.
Introduction
Proteins with unnatural modifications such as drug-conjugated antibodies and protein probes containing fluorophores have attracted much attention in chemical biology and drug development. A chemical reaction often employed for the preparation of modified proteins is native chemical ligation (NCL).1–5) This process produces proteins by peptide bond formation between a peptide or protein with a C-terminal thioester and a peptide or protein with an N-terminal cysteine (Cys). Consequently, peptide thioesters are a key synthetic intermediate for NCL-mediated synthesis and semi-synthesis of proteins. The most popular protocol for preparation of protein thioesters is the first step of expressed protein ligation (EPL).4,5) This protocol utilizes intein-fused recombinant proteins that automatically isomerize to the protein thioester using the self-processing feature of the intein.6,7) Though used widely, the intein-mediated protocol sometimes works poorly due in part to incomplete folding or low solubility of the intein-fused proteins.8) Therefore, other thioesterification protocols that can be used with expressed proteins have been developed. A typical example of alternative thioesterification methods employs a transpeptidase such as sortase A (SrtA)9) or butelase 1.10) A potential disadvantage of these methods is that a peptide sequence, which is required for recognition by the transpeptidase, remains in the resulting protein thioesters. A synthesis that is traceless, i.e., does not leave extra sequences is thus required.11–16)
We recently developed a traceless preparation of peptide/protein thioesters by the use of carboxypeptidase Y (CPaseY) (EC 3.4.16.5),16) which is a commercially available serine protease, easily isolated from baker’s yeast17,18) (Chart 1A). Treatment of a substrate 1 possessing a C-terminal Cys-proline (Pro)-leucine (Leu)-OH (CPL) sequence with CPaseY in the presence of hydrazine and some additives gives the hydrazinolysis product 2 instead of a hydrolyzed product. After activation of the acyl hydrazine unit of 2 to the acyl azide according to the method published by Liu and colleagues,19) the generated intermediate is spontaneously converted via a diketopiperazine formation to a peptide/protein thioester lacking an additional sequence.20,21) CPaseY preferentially hydrolyzes a hydrophobic C-terminal amino acid but not a hydrophilic residue or proline. Consequently, introduction of the Pro-Leu-OH sequence at a C-terminal position of the substrate suppresses further hydrazinolysis (over-reaction) of intermediate 2 because hydrazinolysis of Leu is much faster than that of the subsequent Pro.

After extensive efforts to apply our CPaseY method to various peptides and proteins, we found that the over-reaction products 3 were sometimes generated even if the CPL sequence was fused to a C-terminus of the substrate (Chart 1B). In such cases, a hydrophobic amino acid was present at the X position. Incorporation of a hydrophilic residue at the X position prevented the over-reaction but in such cases conversion of 1 to 2 was suppressed. These observations agree with substrate preference of CPaseY that favors hydrophobic residues at a C-terminal region.22,23) Both over-reaction of 2 and low conversion of 1 reduce a yield of the thioester product; therefore, we sought to develop an X-independent CPaseY protocol.
Results and Discussion
Design of an X-Independent Thioesterification MethodTo prevent the X-induced over-reaction and low conversion, we planned to introduce a spacer between X and the CPL sequence to exclude X from CPaseY’s substrate recognition pocket. Such a spacer must satisfy the following two conditions: 1) it should not remain in a product peptide/protein, otherwise the process would not be traceless; 2) it should give a product peptide/protein even if the over-reaction occurs. A repetitive Cys-Pro (CP) sequence was chosen as the spacer (Chart 2) that could meet these requirements. Treatment of substrate 4, which possesses a multiple CP sequence between X and L, with CPaseY and hydrazine followed by NaNO2 and a thiol would give thioester 6 (m = n − 1). Then, repetitive removal of CP by spontaneous diketopiperazine formation gives the peptide/protein thioester 8 lacking the spacer moiety. If over-reaction of intermediate 5 occurs, the newly generated 7 can also be converted to product 8 via 6 (m = n − 2) as similar to 5.
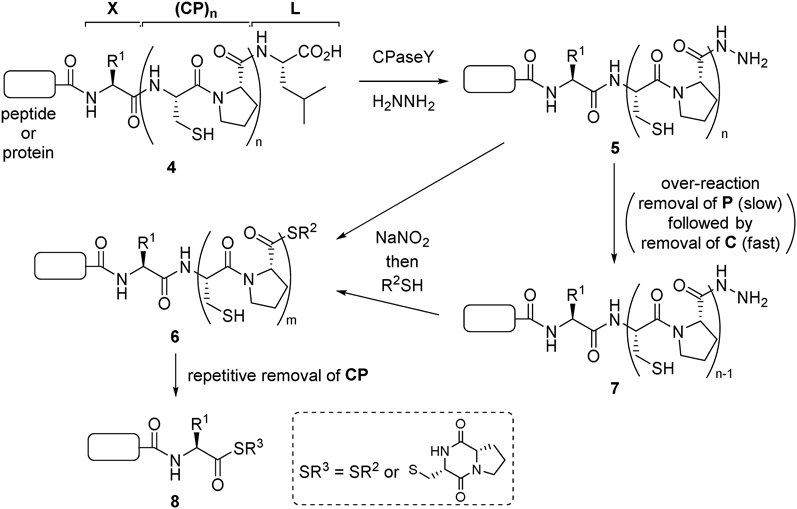
Chart 2. CPaseY-Mediated Traceless Synthesis of Peptide/Protein Thioester Employing a Repeated CP Spacer
The effect of the length of the CP spacer on conversion of a substrate was first examined (Chart 3A). In this research, peptides, not proteins were employed as the substrate because of the simpler structural analysis. Substrate 9 with a varying number of CP sequences (n = 1, 2 or 3) was treated with CPaseY under previously optimized reaction conditions.16) A hydrophobic Phe (F) or a hydrophilic Lys (K) was introduced at the X position. The time course of the conversion of 9 to 10 is summarized in Chart 3B. In the case of the original substrates that do not have a repetitive CP spacer (n = 1), time-dependent reduction of the conversion caused by the over-reaction of 10 was observed for 9 with a hydrophobic F at the X position. When the F was replaced with the hydrophilic K, the reaction was incomplete, with only approx. 25% conversion after 6 h, as mentioned in the Introduction, above. Insertion of an additional CP sequence (n = 2) suppressed the over-reaction (X = F) and improved the conversion (X = K). Finally, X-independent and highly efficient conversion of 9 was achieved by the use of a triple CP sequence (n = 3). We next verified the generality of this triple CP strategy.

To verify the generality of the triple CP strategy, it was applied to two different peptides 11 and 16. Conversion of peptide 11 (n = 1 or 3) to 12 was examined as shown in Chart 4. Introduction of the triple CP sequence successfully suppressed generation of over-reaction byproducts 13 and 14. In the case of 11 (n = 3), a small amount of over-reaction product 12 (n = 2) was detected but causes no problem because 12 (n = 2) can be converted to the corresponding thioester as described in Chart 2. Hydrazinolysis of peptide 16 (n = 1 or 3) is similar to that of 11 and the results were summarized in Chart 5. Although over-reaction products 18 and 19 were generated in a time-dependent fashion in the case of 16 (n = 1), only desired products 17 (n = 2 and 3) were obtained from 16 (n = 3). These results showed that our triple CP strategy generally can suppress the over-reaction and produce the acyl hydrazine product in high purity.

Azidation of an acyl hydrazine, which can be generated by the triple CP strategy, followed by thioesterification and subsequent NCL in one-pot reaction was next examined (Chart 6). Following the azidation of substrate 20 to 23, a counterpart of NCL (22) and 4-mercaptophenylacetic acid (MPAA) were added to the reaction mixture and progress of the reaction was monitored by HPLC. Shortly after addition of the reagents, some intermediate thioesters 24 were detected with NCL product 21 (Chart 6C). It is well-known that NCL of Pro-thioesters is too slow under standard NCL conditions.24–26) Consequently, intermediates 24 (n > 0) should preferentially be involved in removal of the CP units, but not in NCL, and converge to Ala-thioester 24 (n = 0), which can be ligated with 22 to give product 21. Actually, all the intermediates 24 disappeared and the ligated product 21 was obtained in high purity (Chart 6D). These results encouraged us to apply the triple CP strategy to preparation of naturally occurring peptides to demonstrate its practicality.

Application of the triple CP strategy to preparation of the reduced form (rf) Antimicrobial Peptide 1 (AMP1, UniProt KB AC: B3EWQ1) was attempted (Chart 7). Hydrazinolysis of N-terminal fragment 16 by CPaseY was conducted according to the procedure described above, and an acyl hydrazine 17 was obtained in high purity (Chart 7B). Subsequent azidation, thioesterification and NCL with 25 in one-pot manner afforded rf-AMP1 (26) in 60% isolated yield. This result demonstrates that the triple CP strategy should be applicable to the synthesis of peptides/proteins.
Conclusion
A sequence-independent CPaseY-mediated protocol for synthesis of thioesters has been established. An earlier version of the protocol includes the CPaseY-mediated hydrazinolysis of the Pro-Leu junction followed by the auto-processing of the Cys-Pro unit at the C-terminal end of a peptide corresponding to an aminoacyl (X) Cys-Pro-Leu-OH (X-CP-L-OH) sequence to afford X-thioester in a traceless manner. One limitation of the conventional protocol is that the progress of the initial hydrazinolysis step depends on the nature of the X residue. Incomplete or over-hydrazinolysis occurs in the presence of a hydrophilic or hydrophobic X residue, respectively. The insight that such X-dependency is attributable to the properties of the substrate-binding pocket is of great preference to hydrophobic residues prompted us to incorporate additional CP repeats in the C-terminal portion of peptide substrates. Incorporated CP repeats ((CP)n) do not only place X distant from the reaction center but also produce the desired prolyl hydrazide in an X-independent manner. Application of the CPaseY-mediated hydrazinolysis to substrates with triple CP repeats, X-(CP)3-L-OH at the C-terminal end, efficiently affords the corresponding X-(CP)3-NHNH2 which was then converted to the X thioester with the triple CP repeat auto-processed. Finally, the utility of the developed triple CP strategy in the traceless preparation of thioesters was demonstrated by the synthesis of a 23-residue antimicrobial peptide. Further application of the triple CP strategy to recombinant protein is underway in our laboratory and will be reported in due course.
Experimental
General MethodsMass spectra were recorded on a Waters MICROMASS® LCT PREMIER™ (electrospray ionization-time-of-flight (ESI-TOF)) or a Shimadzu LCMS-2020 (ESI-Q). For HPLC, separation was achieved with a Cosmosil 5C18-AR-II column (Nacalai Tesque, Japan; analytical: 4.6 × 250 mm, flow rate 1.0 mL/min; semi-preparative: 10 × 250 mm, flow rate 3.0 mL/min; preparative: 20 × 250 mm, flow rate 10.0 mL/min), and eluants were detected by UV at 220 nm or by MS. A solvent system consisting of 0.1% (v/v) trifluoroacetic acid (TFA) aqueous solution (solvent A) and 0.1% (v/v) TFA in acetonitrile (solvent B) was used for HPLC elution. Characterization data of peptides are summarized in Table 1.
Table 1. Characterization Data of Peptides
| Comp. # | Yield (%) | HPLC | MS (m/z) |
|---|
| Analytical | p or spa) |
|---|
| Gradientb) (%) | Retention time (min) | Gradientb) (%) | Calcd | Found |
|---|
| 9 (X = F, n = 1) | 49 | 5–40 | 25.5 | 22–32 (p) | 982.5 [M + H]+ | 982.6 |
| 9 (X = K, n = 1) | 35 | 5–30 | 23.5 | 12–22 (p) | 963.5 [M + H]+ | 963.6 |
| 9 (X = F, n = 2) | 31 | 5–70 | 17.0 | 20–30 (sp) | 1182.6 [M + H]+ | 1182.5 |
| 9 (X = K, n = 2) | 45 | 5–35 | 23.2 | 10–20 (sp) | 1163.6 [M + H]+ | 1163.6 |
| 9 (X = F, n = 3) | 36 | 5–60 | 19.6 | 15–25 (sp) | 1382.6 [M + H]+ | 1382.5 |
| 9 (X = K, n = 3) | 21 | 5–35 | 24.9 | 5–15 (sp) | 1363.6 [M + H]+ | 1363.6 |
| 10 (X = F, n = 1) | — | 5–35 | 19.9 | — | 883.5 [M + H]+ | 883.4 |
| 10 (X = K, n = 1) | — | 5–35 | 13.3 | — | 864.5 [M + H]+ | 864.4 |
| 10 (X = F, n = 2) | — | 5–60 | 14.6 | — | 1083.5 [M + H]+ | 1083.4 |
| 10 (X = K, n = 2) | — | 5–35 | 15.6 | — | 1064.5 [M + H]+ | 1064.5 |
| 10 (X = F, n = 3) | — | 5–60 | 15.4 | — | 1283.6 [M + H]+ | 1283.8 |
| 10 (X = K, n = 3) | — | 5–35 | 17.6 | — | 1264.6 [M + H]+ | 1264.9 |
| 11 (n = 1) | 6 | 5–60 | 19.9 | 25–35 (sp) | 1539.8 [M + H]+ | 1540.1 |
| 11 (n = 3) | 17 | 5–60 | 20.5 | 21–31 (sp) | 970.5 [M + 2H]2+ | 970.4 |
| 12 (n = 1) | — | 5–60 | 15.4 | — | 720.9 [M + 2H]2+ | 720.9 |
| 12 (n = 2) | — | 5–60 | 16.3 | — | 820.9 [M + 2H]2+ | 820.8 |
| 12 (n = 3) | — | 5–60 | 16.9 | — | 921.0 [M + 2H]2+ | 920.8 |
| 13 | — | 5–60 | 11.7 | — | 1093.6 [M + H]+ | 1093.7 |
| 14 | — | 5–60 | 9.5 | — | 980.6 [M + H]+ | 980.6 |
| 15 | — | 5–60 | 16.0 | — | 960.2 [M + 2H]3+ | 960.0 |
| 16 (n = 1) | 11 | 5–60 | 19.4 | 20–30 (sp) | 989.5 [M + 2H]2+ | 989.3 |
| 16 (n = 3) | 43 | 5–60 | 20.2 | 24–34 (p) | 1189.6 [M + 2H]2+ | 1189.3 |
| 17 (n = 1) | — | 5–60 | 15.5 | — | 940.0 [M + 2H]2+ | 940.1 |
| 17 (n = 2) | — | 5–60 | 16.3 | — | 1040.0 [M + 2H]2+ | 1040.0 |
| 17 (n = 3) | 74 | 5–60 | 16.8 | 15–25 (p) | 760.3 [M + 3H]3+ | 760.5 |
| 18 | — | 5–60 | 11.3 | — | 716.9 [M + 2H]2+ | 716.8 |
| 19 | — | 5–60 | 12.9 | — | 709.9 [M + 2H]2+ | 709.7 |
| 20 | 1 | 5–35 | 18.7 | 8–18 (p) | 1207.6 [M + H]+ | 1208.0 |
| 21 | — | 5–30 | 20.5 | — | 592.8 [M + 2H]2+ | 592.7 |
| 22 | 6 | 5–30 | 12.3 | 5–15 (p) | 609.3 [M + H]+ | 609.3 |
| 24 (DKP, n = 0) | — | 5–30 | 17.8 | — | 775.4 [M + H]+ | 775.6 |
| 24 (DKP, n = 1) | — | 5–30 | 22.4 | — | 975.5 [M + H]+ | 975.8 |
| 24 (DKP, n = 2) | — | 5–30 | 25.1 | — | 588.3 [M + 2H]2+ | 588.5 |
| 24 (Ar, n = 0) | — | 5–30 | 26.3 | — | 743.4 [M + H]+ | 743.6 |
| 25 | 12 | 5–60 | 13.9 | 11–21 (p) | 901.1 [M + 3H]3+ | 901.3 |
| 26 | 60 | 5–60 | 16.9 | 15–25 (p) | 1449.7 [M + 3H]3+ | 1449.8 |
a) p: preparative; sp: semi-preparative. b) Gradient over 30 min.
Side chain protecting groups of Fmoc amino acids are as follows: tert-butoxycarbonyl (Boc): K; OtBu: D and E; Pbf: R; tBu: S, T, and Y; Trt: C, N, and Q. Elongations of peptides were achieved by repetition of the coupling reaction and the Fmoc removal as described below.
Coupling reaction: According to a protocol shown in Table 2, an Fmoc amino acid (4.0 equivalent (eq.)) in N,N-dimethylformamide (DMF) was coupled on a resin at r.t. for 2 h. Completion of the coupling was checked by the Kaiser ninhydrin test. The coupling reaction is repeated until the Kaiser test became negative. After completion of the coupling, the resin obtained was washed 5 times with DMF.
Table 2. Coupling Protocols
| Protocol | Reagents (abbreviation and/or eq.) |
|---|
| 1 | N,N′-Diisopropylcarbodiimide (DIPCI, 4.0 eq.), 1-hydroxybenzotriazole monohydrate (HOBt·H2O, 4.0 eq.) |
| 2 | O-(1H-Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, 3.9 eq.), N,N-diisopropylethylaminie (DIPEA, 4.0 eq.) |
| 3 | O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, 3.9 eq.), DIPEA (4.0 eq.) |
Removal of Fmoc groups: A resin was treated with 20% (v/v) piperidine in DMF at r.t. for 10 min, and then washed 10 times with DMF.
Release of deprotected peptide from resin: After treatment of a resin with a cleavage cocktail, (TFA/m-cresol/thioanisole/1,2-ethanedithiol/H2O/triisopropylsilane (80/5/5/5/-2.5/2.5 (v/v)), 100 µL/1 mg resin) at r.t. for 2 h, the resin was filtered off and the filtrate was concentrated in an N2 stream. Cooled diethyl ether (Et2O) was added to the concentrate and the precipitate was collected by centrifugation. The obtained precipitate was thoroughly washed with cooled Et2O, and the peptide in the precipitate was purified by preparative HPLC. The eluted solution of the product was finally lyophilized to give the peptide as a powder.
Preparation of Peptides 9, 11, 16, 22, and 25Typical procedure: N-Fmoc-L-Leu (10 eq.) was loaded on Wang resin (0.80 mmol/g) with the aid of HBTU (9.9 eq.), N,N-diisopropylethylamine (DIPEA) (10 eq.) and N,N-dimethylaminopyridine (DMAP, 0.05 eq.) in DMF at r.t. for 12 h. The resulting resin was then treated with Ac2O (10 eq.) and pyridine (10 eq.) in DMF at r.t. for 30 min. The amount of leucine that was loaded was estimated based on the level of the Fmoc group on the resin. On the leucine-loaded Wang resin, peptides 9 were synthesized according to the General Procedure for Fmoc SPPS. For preparation of 16, 22 and 25, HMPB-ChemMatrix resin (for 16 and 25, 0.50 mmol/g) and NovaSyn® TGR resin (for 22, 0.25 mmol/g) were employed instead of Wang resin, respectively.
Preparation of Hydrazine Peptide 20 by SPPS19)P-Nitrophenyl chloroformate (3.0 eq.) was added at 0 °C to Wang resin (0.80 mmol/g) and N-methylmorpholine (3.0 eq.) in CH2Cl2, and the resulting mixture was shaken at r.t. for 24 h. After filtration, the resin that was obtained was washed with CH2Cl2, DMF, MeOH and then CH2Cl2. H2NNH2·H2O (7.0 eq.) was added to the resulting resin in DMF/CH2Cl2 (4 : 2 (v/v)) at 0 °C, and the mixture was shaken at r.t. for 21 h. Following to filtration and washing with CH2CH2, DMF, MeOH and subsequent CH2Cl2, peptide 20 was synthesized on the obtained hydrazine carboxylate Wang resin according to the General Procedure for Fmoc SPPS.
Hydrazinolysis of Peptides 9, 11 and 16 Using CPaseYTypical procedure: As described in the previous paper,16) peptide 9 (0.050 µmol) in 50 µL aqueous solution with 0.1 µM CPaseY, 0.2 M H2NNH2·H2O and 60 mM cyclohexanone was incubated at 25 °C (pH 6.4). Before HPLC analysis, an aqueous solution of 6 M Gn·HCl, 200 mM MeONH2·HCl and 200 mM tris(2-carboxyethyl)-phosphine (TCEP)·HCl was added twice to the reaction mixture, and the resulting mixture was incubated at 37 °C for 10 min.
Azidation of 20 Followed by Thioesterification and Subsequent NCL with 22Typical procedure: An aqueous solution of NaNO2 (0.1 M, 2.5 µL) was added at −10 °C to a solution of 20 (0.050 µmol) in 50 mM sodium phosphate buffer with 6 M Gn·HCl (pH 3.0, 32 µL) and the mixture was stored at −10 °C for 30 min. Then 50 mM sodium phosphate buffer (50 mM, 17 µL) containing 6 M Gn·HCl and 150 mM MPAA was added to the reaction mixture, and its pH was adjusted to 6.5 with 1.0 M NaOH aqueous solution. Following addition of 22 (0.10 µmol) and subsequent shaking at r.t. for 3 h, the reaction mixture was analyzed by LC/MS after quenching twice with an aqueous solution of Gn·HCl (6 M) and TCEP·HCl (200 mM) at 37 °C for 10 min.
Synthesis of rf-AMP1 (26)Starting from 16 (1.0 µmol), the hydrazinolysis product 17 (n = 3) was obtained as described in “Hydrazinolysis of peptides 9, 11 and 16 using CPaseY” in 74% yield (1.7 mg, 0.74 µmol). The obtained hydrazinolysis product 17 (n = 3) was subjected to azidation, thioesterification, and NCL with 25 as in treatment of 20, and 2.6 mg of rf-AMP1 (26, 0.60 µmol) was obtained in 60% yield.
Acknowledgments
This research was supported in part by Grants-in-Aid for Scientific Research (KAKENHI, 16H02611 for A. O. and 20K21483 for A. S.) and by The Canon Foundation (for A. O.). A. S. is grateful for support from the Research Foundation for Pharmaceutical Sciences.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B. H., Science, 266, 776–779 (1994).
- 2) Kent S. B. H., Chem. Soc. Rev., 38, 338–351 (2009).
- 3) Kulkarni S. S., Sayers J., Premdjee B., Payne R. J., Nat. Rev. Chem., 2, 122 (2018).
- 4) Muir T. W., Annu. Rev. Biochem., 72, 249–289 (2003).
- 5) Vila-Perello´ M., Muir T. W., Cell, 143, 191–200 (2010).
- 6) Severinov K., Muir T. W., J. Biol. Chem., 273, 16205–16209 (1998).
- 7) Muir T. W., Sondhi D., Cole P. A., Proc. Natl. Acad. Sci. U.S.A., 95, 6705–6710 (1998).
- 8) Valiyaveetil F. I., Mackinnon R., Muir T. W., J. Am. Chem. Soc., 124, 9113–9120 (2002).
- 9) Ling J. J., Policarpo R. L., Rabideau A. E., Liao X., Pentelute B. L., J. Am. Chem. Soc., 134, 10749–10752 (2012).
- 10) Cao Y., Nguyen G. K. T., Tam J. P., Liu C.-F., Chem. Commun., 51, 17289–17292 (2015).
- 11) Okamoto R., Morooka K., Kajihara Y., Angew. Chem. Int. Ed., 51, 191–196 (2012).
- 12) Adams A. L., Cowper B., Morgan R. E., Premdjee B., Caddick S., Macmillan D., Angew. Chem. Int. Ed., 52, 13062–13066 (2013).
- 13) Kajihara Y., Kanemitsu Y., Nishihara M., Okamoto R., Izumi M., J. Pept. Sci., 20, 958–963 (2014).
- 14) Tsuda Y., Shiganaga A., Tsuji K., Denda M., Sato K., Kitakaze K., Nakamura T., Inokuma T., Itoh K., Otaka A., ChemistryOpen, 4, 448–452 (2015).
- 15) Miyajima R., Tsuda Y., Inokuma T., Shiganaga A., Imanishi M., Futaki S., Otaka A., Pept. Sci., 4, 531–546 (2016).
- 16) Komiya C., Shigenaga A., Tsukimoto J., Ueda M., Morisaki T., Inokuma T., Itoh K., Otaka A., Chem. Commun., 55, 7029–7032 (2019).
- 17) Hayashi R., Bai Y., Hata T., J. Biochem., 77, 1313–1318 (1975).
- 18) Endrizzi J. A., Breddam K., Remington S. J., Biochemistry, 33, 11106–11120 (1994).
- 19) Fang G.-M., Li Y.-M., Shen F., Huang Y.-C., Li J.-B., Lin Y., Cui H.-K., Liu L., Angew. Chem. Int. Ed., 50, 7645–7649 (2011).
- 20) Kawakami T., Aimoto S., Chem. Lett., 36, 76–77 (2007).
- 21) Kawakami T., Shimizu S., Aimoto S., Bull. Chem. Soc. Jpn., 83, 570–574 (2010).
- 22) Nakase H., Murata S., Ueno H., Hayashi R., Biosci. Biotechnol. Biochem., 65, 2465–2471 (2001).
- 23) Hecht K. A., O’Donnell A. F., Brodsky J. L., Cell. Logist., 4, e28023 (2014).
- 24) Hackeng T. M., Griffin J. H., Dawson P. E., Proc. Natl. Acad. Sci. U.S.A., 96, 10068–10073 (1999).
- 25) Pollock S. B., Kent S. B. H., Chem. Commun., 47, 2342–2344 (2011).
- 26) Choudhary A., Fry C. G., Kamer K. J., Raines R. T., Chem. Commun., 49, 8166–8168 (2013).


