Abstract
A turn-on fluorescent traceable linker based on N-sulfanylethylcoumarinyl amide (SECmide) has been developed as an advanced cleavable linker. It was successfully employed for the enrichment and selective visualization of a target protein in cell lysate. The results demonstrated that the SECmide-based traceable linker is potentially applicable to the identification of low molecular weight target proteins, a factor which has been problematic for a previously developed N-sulfanylethylanilide-based traceable linker.
Introduction
Many bioactive ligands represented by natural products and active pharmaceutical ingredients exhibit their biological activities through specific interactions with target proteins such as receptors, ion channels and enzymes. Therefore, the identification of unknown target proteins interacting with the ligands is indispensable for understanding their biological mechanisms of action and for drug development. Tag-based target identification is widely utilized in this field, and a commonly used method is as follows. An alkyne-containing ligand derivative is first covalently connected to the target protein by photo-affinity labeling1) or an activity-based probe approach.2–5) To the resulting target with an alkyne moiety is then attached a linker possessing a biotin moiety and an azide via Cu(I)-catalyzed alkyne–azide cycloaddition (CuAAC).6–9) Next, the target cross-linked with the biotinylated linker is selectively adsorbed on streptavidin (SAv) beads.10,11) A final step before sequence analyses is the elution of the enriched target from the SAv beads; however, this step is quite problematic. Elution efficiency of the target protein under mild conditions is typically low due to the high affinity of the SAv-biotin interaction.12) Efficient elution of the target could be achieved with the use of harsh conditions; however, contamination of nonspecifically adsorbed non-target proteins is generally unavoidable. To overcome these problems, cleavable linkers, in which a biotin moiety and azide are connected by a unit cleavable under mild conditions, have been invented.13–16) These cleavable linkers usually allow for the efficient elution of the target without contamination of non-targets upon its cleavage. However, it was reported that the contamination of non-targets could sometimes not be prevented, even with the use of the cleavable linker.17–19) Because the contamination of non-target proteins hampers the subsequent identification of target proteins, several research groups including ours were prompted to engage in the development of an advanced cleavable linker.20–26)
The new cleavable linkers were designed to selectively label a target protein for facile discrimination of the target from non-targets. These linkers enable label-based tracing of the target; therefore, we named this type of cleavable linker a “traceable linker.” We previously developed an N-sulfanylethylanilide (SEAlide)-based traceable linker (1) as shown in Chart 1A.20,27) Traceable linker 1 is first connected with an alkynylated target protein by CuAAC, and the generated conjugate is then adsorbed on SAv beads, similar to the case of the cleavable linkers. Because phosphate activates the SEAlide unit from an amide to a thioester form,28,29) the addition of a phosphate buffer containing reductant for the removal of a t-BuS group of the SEAlide moiety was necessary, and a labeling reagent with a cysteine moiety induced bioothogonal Native Chemical Ligation (NCL)30) to elute a labeled target protein. In this protocol, an external labeling reagent is required, similar to the other traceable linkers,21–25) and complete removal of the excess labeling reagent was sometimes complicated. When sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequent fluorescence imaging of the eluent with the residual labeling reagent was examined, fluorescence caused by the labeling reagent was observed in the low molecular weight region.31) This fluorescence hampers label-based identification of the target proteins if its molecular weight is low; therefore, we decided to re-design traceable linker 1. Herein, we report a new traceable linker that does not require external labeling reagents.
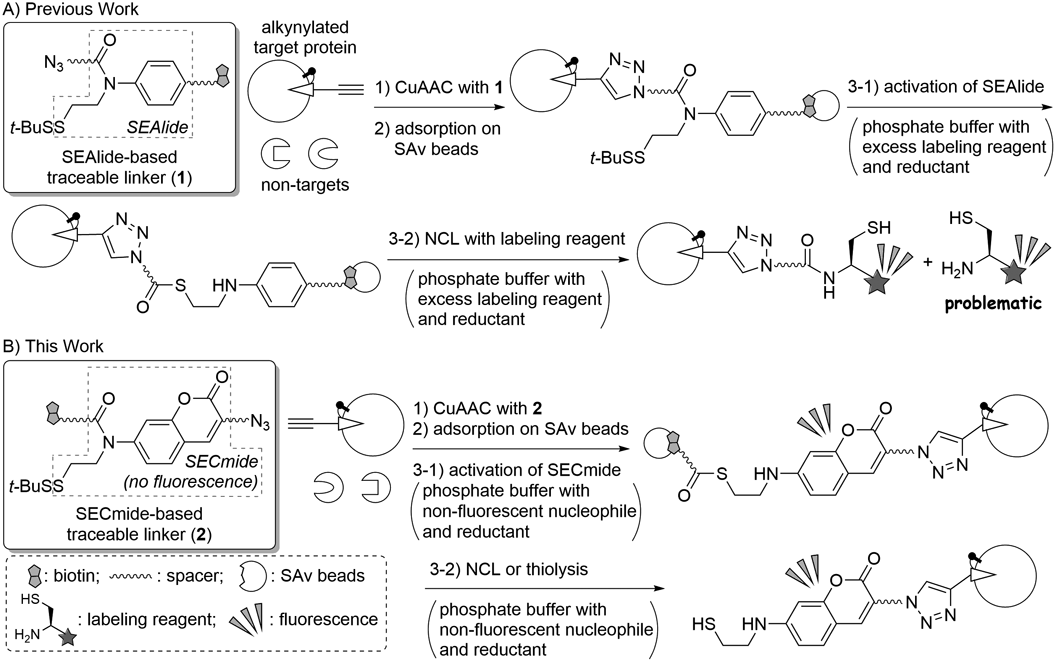
Results and Discussion
In this study, we designed turn-on fluorescent traceable linker 2 composed of N-sulfanylethylcoumarinyl amide (SECmide)32) (Chart 1B). The SECmide-based traceable linker 2 is not fluorescent because its coumarinyl amine is acylated. It could be introduced onto the alkynylated target protein, similarly to traceable linker 1. After adsorption on SAv beads, treatment with phosphate buffer containing a non-fluorescent nucleophile and a reductant would induce the elution of the target via removal of the t-BuS group, activation of the SECmide moiety to the corresponding thioester form, and subsequent nucleophilic cleavage of the thioester via NCL or a thiolysis reaction. The eluted target has an aminocoumarin moiety without N-acylation: therefore, the target protein can be fluorescently visualized without the use of external labeling reagents.
SECmide-based traceable linker 3 shown in Fig. 1 was synthesized using Fmoc-based solid phase peptide synthesis (Fmoc SPPS) (Chart S1). CuAAC, adsorption on SAv beads, and elution from the beads were next examined using a purified alkynylated protein. CuAAC of traceable linker 3 with an alkynylated bovine serum albumin (BSA),21) which is widely employed as a model alkynylated target protein, was conducted. The CuAAC afforded the traceable linker-BSA conjugate possessing a biotin moiety, as shown in Fig. 2A (lane 2). The CuAAC product was then adsorbed on SAv beads in Tris buffered saline (TBS) with SDS. Following washing with TBS, the resulting beads were treated with 0.4 M phosphate buffer (pH 7.0) containing 100 µM cysteine as a nucleophile, 50 mM 4-mercaptophenylacetic acid (MPAA) as a catalyst for NCL of the SECmide moiety, 40 mM tris(2-carboxyethyl)phosphine (TCEP) as a reductant, and 0.1% (w/v) SDS for elution of the protein via linker cleavage. Finally, SDS-PAGE followed by fluorescence imaging of the obtained eluent demonstrated that the BSA with a fluorophore was successfully eluted, as shown in Fig. 2B (lane 1). The elution conditions described above employed a cocktail of many reagents including MPAA, TCEP, and cysteine. Key requirements for widely used methods are operational simplicity and low cost. The use of a DTT alternate to MPAA and cysteine (for linker cleavage mediated by NCL) and TCEP (for removal of t-BuS group) was likely to fulfill these requirements. That is, cost-effective DTT not only participates in the thiolytic linker cleavage reaction but also works as the reagent for the reductive removal of the t-BuS group. Results of DTT-induced elution are shown in Fig. 2B (lanes 3 and 4). Efficiency of the elution was comparable to that of NCL (lanes 1 and 2). We next attempted to dilute phosphate because a high concentration of phosphate sometimes causes salting out of proteins to reduce the elution efficiency.33) The results unexpectedly indicated that the elution efficiency decreased as the concentration of phosphate decreased (Fig. 2C). This can be explained by inefficiency of the linker cleavage because phosphate catalyzes the activation of the SECmide moiety to the corresponding thioester form.32) We therefore utilized 0.4 M phosphate buffer, which is the densest phosphate buffer we examined in this study, for the following DTT-induced elution.
To demonstrate the necessity of activation of the SECmide moiety to the thioester form, S-methylated negative control 4 was also tested (Fig. 1). The alkynylated BSA was connected with 4 by CuAAC (Fig. 2A, lane 3),34) and the obtained conjugate was adsorbed on SAv beads. The obtained beads were then treated with DTT in phosphate buffer, and the eluent and proteins remaining on the beads were analyzed (Fig. 2D). In this case, elution of the BSA conjugate was prevented. These results indicate that the linker cleavage occurred via the thioester form, as we designed.
Finally, we applied traceable linker 3 to the enrichment and selective visualization of the alkynylated target protein in a cell lysate. In this study, a mixture of the alkynylated BSA in a lysate of HeLa cells was employed (Fig. 3, CBB staining, lane 3). After CuAAC of the mixture with traceable linker 3, followed by adsorption on SAv beads, the proteins were eluted by treatment with 200 mM DTT in 0.4 M phosphate buffer with 0.1% (w/v) SDS. As shown in Fig. 3 (CBB staining, lane 4), the target BSA conjugate was successfully enriched. In this case, it was accompanied by a small amount of contaminant proteins. However, discrimination of the target from the non-targets was achieved based on fluorescence imaging (Fig. 3, fluorescence imaging, lane 4). Furthermore, there was no background fluorescence at a low molecular weight region because of non-use of excess labeling reagents. This suggests that the turn-on fluorescent traceable linker 3 is potentially applicable to the identification of low molecular weight target proteins.
In conclusion, turn-on fluorescent traceable linker 3 based on the SECmide was developed. Whereas previously reported traceable linkers require the use of external labeling reagents that complicate the detection of low molecular weight target proteins, the new traceable linker invented in this study does not require labeling reagents for its visualization. We believe that such an unprecedented feature should contribute to the easy identification of low molecular weight target proteins in the near future.
Acknowledgments
The authors are grateful to Professors Kohji Itoh and Daisuke Tsuji (Tokushima University) for their support in preparing a lysate of HeLa cells. This research was supported in part by a Grant-in-Aid for Scientific Research (KAKENHI, 16H02611 for A.O. and 18J13939 for T.M.).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
References
- 1) Chen Y., Topp E. M., J. Pharm. Sci., 108, 791–797 (2019).
- 2) Kubota K., Funabashi M., Ogura Y., Biochim. Biophys. Acta Proteins Proteomics, 1867, 22–27 (2019).
- 3) Li N., Overkleeft H. S., Florea B. I., Curr. Opin. Chem. Biol., 16, 227–233 (2012).
- 4) Wang K., Yang T., Wu Q., Zhao X., Nice E. C., Huang C., Expert Rev. Proteomics, 9, 293–310 (2012).
- 5) Cravatt B. F., Wright A. T., Kozarich J. W., Annu. Rev. Biochem., 77, 383–414 (2008).
- 6) Mandoli A., Molecules, 21, 1174 (2016).
- 7) Hein J. E., Fokin V. V., Chem. Soc. Rev., 39, 1302–1315 (2010).
- 8) Tornøe C. W., Christensen C., Meldal M., J. Org. Chem., 67, 3057–3064 (2002).
- 9) Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed., 41, 2596–2599 (2002).
- 10) Savage M. D., BioMethods, 7, 1–29 (1996).
- 11) Hofmann K., Kiso Y., Proc. Natl. Acad. Sci. U.S.A., 73, 3516–3518 (1976).
- 12) Green N. M., Adv. Protein Chem., 29, 85–133 (1975).
- 13) Yang Y., Fonovic M., Verhelst S. H. L., Methods Mol. Biol., 1491, 185–203 (2017).
- 14) Bielski R., Witczak Z., Chem. Rev., 113, 2205–2243 (2013).
- 15) Rudolf G. C., Heydenreuter W., Sieber S. A., Curr. Opin. Chem. Biol., 17, 110–117 (2013).
- 16) Leriche G., Chisholm L., Wagner A., Bioorg. Med. Chem., 20, 571–582 (2012).
- 17) Verhelst S. H. L., Fonović M., Bogyo M., Angew. Chem. Int. Ed., 46, 1284–1286 (2007).
- 18) Paulick M. G., Hart K. M., Brinner K. M., Tjandra M., Charych D. H., Zuckermann R. N., J. Comb. Chem., 8, 417–426 (2006).
- 19) van der Veken P., Dirksen E. H. C., Ruijter E., Elgersma R. C., Heck A. J. R., Rijkers D. T. S., Slijper M., Liskamp P. M. J., ChemBioChem, 6, 2271–2280 (2005).
- 20) Morisaki T., Denda M., Yamamoto J., Tsuji D., Inokuma T., Itoh K., Shigenaga A., Otaka A., Chem. Commun., 52, 6911–6913 (2016).
- 21) Lee S., Wang W., Lee Y., Sampson N. S., Org. Biomol. Chem., 13, 8445–8452 (2015).
- 22) Yamamoto J., Denda M., Maeda N., Kita M., Komiya C., Tanaka T., Nomura W., Tamamura H., Sato Y., Yamauchi A., Shigenaga A., Otaka A., Org. Biomol. Chem., 12, 3821–3826 (2014).
- 23) Yamamoto J., Maeda N., Komiya C., Tanaka T., Denda M., Ebisuno K., Nomura W., Tamamura H., Sato Y., Yamauchi A., Shigenaga A., Otaka A., Tetrahedron, 70, 5122–5127 (2014).
- 24) Dirksen A., Yegneswaran S., Dawson P. E., Angew. Chem. Int. Ed., 49, 2023–2027 (2010).
- 25) Park K. D., Liu R., Kohn H., Chem. Biol., 16, 763–772 (2009).
- 26) Shigenaga A., Chem. Pharm. Bull., 67, 1171–1178 (2019).
- 27) Hayashi R., Morimoto S., Tomohiro T., Chem. Asian J., 14, 3145–3148 (2019).
- 28) Otaka A., Sato K., Shigenaga A., Top. Curr. Chem., 363, 33–56 (2015).
- 29) Otaka A., Sato K., Ding H., Shigenaga A., Chem. Rec., 12, 479–490 (2012).
- 30) Kent S. B. H., Chem. Soc. Rev., 38, 338–351 (2009).
- 31) See the bottom part of the fluorescence imaging of Fig. S7 in ref. 20.
- 32) Eto M., Naruse N., Morimoto K., Yamaoka K., Sato K., Tsuji K., Inokuma T., Shigenaga A., Otaka A., Org. Lett., 18, 4416–4419 (2016).
- 33) Zhang Y., Cremer P. S., Curr. Opin. Chem. Biol., 10, 658–663 (2006).
- 34) In Fig. 2A, the band intensity of lane 2 of Western blotting was lower than that of lane 3, presumably due to the thermal activation of linker 3 to the thioester form, followed by thiolytic removal of the biotin moiety during pretreatment for SDS-PAGE (reaction conditions: 100°C, 5 min in sample loading buffer that contains mercaptoethanol). It is not problematic for enrichment and labeling of target proteins because SDS-PAGE of a target protein–biotin conjugate is not involved in a protocol for the target enrichment and labeling.


