Abstract
The emergence of multidrug-resistant (MDR) Gram-negative bacteria has become a global problem. Among MDR Gram-negative bacteria, carbapenem-resistant Enterobacteriaceae (CRE), MDR Pseudomonas aeruginosa, and MDR Acinetobacter baumannii have limited treatment options and present serious threats. Therefore, strong countermeasures must be taken against these bacteria immediately. Accordingly, the focus of this review is on recent advances in the development of promising antibacterial agents against MDR Gram-negative bacteria. These agents include novel tetracyclines, polymyxins, β-lactams, β-lactam/β-lactamase inhibitors, aminoglycosides, and peptide mimetics that have been recently approved or have shown promising results in clinical and preclinical development. This review summarizes these potent antibiotics in terms of their development status, mode of action, spectra of activity, and structure–activity relationship.
1. Introduction
The emergence of multidrug-resistant (MDR) Gram-negative bacteria has become a global concern.1–3) Accordingly, MDR Gram-negative bacteria have been defined as problematic pathogenic bacteria by the United States Centers for Disease Control and Prevention (CDC) and by WHO. In 2013, the United States CDC declared that carbapenem-resistant Enterobacteriaceae (CRE),4) as well as drug-resistant Neisseria gonorrhoeae and Clostridium difficile are an urgent threat to public health.5) Furthermore, WHO recently published its “priority list of antibiotic-resistant bacteria for R&D.”6) This list classified carbapenem-resistant Acinetobacter baumannii, carbapenem-resistant Pseudomonas aeruginosa,7) and carbapenem-resistant extended spectrum β-lactamase (ESBL)-producing Enterobacteriaceae as “Priority 1: critical.”8,9) This list demonstrates that the development of antibiotics active against MDR Gram-negative bacteria, particularly carbapenemase-producing organisms, is key to preventing future catastrophic pandemic outbreaks. Therefore, strong countermeasures must be taken against these bacteria immediately.
Carbapenemases are enzymes that break down carbapenems via hydrolysis of the β-lactam ring. Of these enzymes, Klebsiella pneumoniae carbapenemase (KPC) and the New Delhi metallo-β-lactamase-1 (NDM-1) are the most problematic enzymes.10) To make matters worse, carbapenemase-producing organisms frequently exhibit resistance mechanisms toward common antibiotics, including fluoroquinolones, tetracyclines, and aminoglycosides. Consequently, there are limited treatment options for carbapenemase-producing organisms.11) This situation has led to the reintroduction in clinical practice of the old and potentially toxic antibiotic colistin as a last resort for the treatment of Enterobacteriaceae-producing KPC and NDM-1.12,13)
To combat MDR Gram-negative bacteria, innovative approaches, including the rational design of drug structures and the discovery of novel mechanisms of action, are desperately needed. As of June 2019, approximately 42 new antibiotics were in clinical development.14,15) Of these, one-third exhibit potent antibacterial activity against any of CRE, carbapenem-resistant A. baumannii, and carbapenem-resistant P. aeruginosa.
Recently, several excellent reviews of antibiotics have been published, including those on antibiotics in the late clinical pipeline and in development,16,17) on treatment options for MDR Gram-negative bacteria,18) and on the strategic optimization of structure.19,20) This review summarizes current potent antibiotics active against dangerous Gram-negative bacteria in terms of their development status, mode of action, spectra of activity, and structure–activity relationship (SAR).
2. Novel Tetracycline-class Antibiotics for MDR Gram-negative Bacteria
Tetracycline antibiotics derived from natural products do not contain substituents at the C-7, C-8, and C-9 positions. However, the development of totally synthetic methods to synthesize tetracyclines has made it possible to modify these three positions,21,22) providing novel and fully synthetic tetracyclines such as XERAVA and TP-6076, developed by Tetraphase Pharmaceuticals Inc. (Fig. 1). The introduction of an electron-withdrawing group at the C-7 position, and substitution at the C-9 position, significantly improved the antimicrobial activity of these synthetic tetracyclines.23,24)
XERAVA (eravacycline, TP-434)25–27) was the first fully synthetic fluorocycline produced by Michael-Dieckmann reaction.28) XERAVA was approved by the U.S. Food and Drug Administration (FDA) in August 27, 2018 for the treatment of complicated intra-abdominal infections (cIAIs) in adults.29–31) XERAVA exhibits broad-spectrum activity against Gram-positive, Gram-negative, and anaerobic bacteria that have acquired tetracycline-specific efflux and ribosomal protection mechanisms.32,33) Furthermore, XERAVA demonstrates potency against MDR bacteria producing ESBL, carbapenemase, and A. baumannii.34–37) In vitro antimicrobial tests demonstrated that XERAVA is 2- to 4-fold more potent than tigecycline38) against Gram-positive and Gram-negative bacteria. The in vivo efficacy of XERAVA utilizing murine thigh infection models was established.39–41)
TP-6076 is currently being evaluated in phase I trials. SAR studies of the C-4, C-7, and C-8 positions demonstrated that electron-withdrawing CF3 and OCF3 groups at the C-7 position resulted in appreciable activity.42,43) In the case of the C-8 position, pyrrolidine resulted in better activity than 4- or 6-membered cyclic amines or N-Me pyrrolidine. Finally, the C-4 position tended to prefer small aliphatic tertiary amines, with the diethylamino analog showing the best antimicrobial activity.44) TP-6076 inhibits bacterial protein synthesis by binding to the 30S ribosomal subunit, and retains activity against Enterobacteriaceae and A. baumannii expressing tetracycline-specific resistance.45) Furthermore, it is unaffected by transferable resistance mechanisms such as KPC, metallo β-lactamase (MBL), OXA, and RNA methylase.46)
3. Novel Polymyxin-class Antibiotics for MDR Gram-negative Bacteria
Polymyxins are cationic lipopeptides and represent a last resort for the treatment of serious MDR Gram-negative bacteria.47) However, their clinical use is limited by their high nephrotoxicity and neurotoxicity. SPR206 and SPR741 (Fig. 2) are novel polymyxin-B derivatives developed by Spero Therapeutics. SPR206 exhibits potency against all ESKAPE (Klebsiella pneumoniae, A. baumannii, P. aeruginosa, or Enterobacter species) pathogens,48) including serine-CRE, metallo-CRE, carbapenem-resistant P. aeruginosa, and A. baumannii.49,50) SPR206 interacts with lipopolysaccharides to disrupt the outer membrane of Gram-negative bacteria.51) The SAR study demonstrated that the position of the amino group in the side chain affected antimicrobial activity and cytotoxicity.52,53) Replacement of the side chain of polymyxin-B with an aminobutyrate unit improved the safety and tolerability of SPR206 compared with those of colistin.54) SPR206 is being evaluated in phase I clinical trials and has acquired qualified infectious disease product (QIDP) status.
SPR741 (formerly NAB741) was designed to minimize the nephrotoxicity of the polymyxin group, and it has completed phase Ia and Ib trials. SPR741 retains the ability to permeate the outer membranes of Gram-negative bacteria. Furthermore, its safety profile is improved compared to that of polymyxin-B.55,56) Development of a drug to treat Gram-negative bacteria has previously been obstructed by the inability to penetrate the bacterial outer membrane. However, this problem has been mitigated by using a potentiator agent that permeabilizes the outer cell membrane. SPR741 potentiates the activity of co-administered antibiotics against Gram-negative bacteria,57,58) although it does not itself have any significant antimicrobial activity.51,59–61)
4. Novel β-Lactams for MDR Gram-negative Bacteria
Cefiderocol (S-649266) (Fig. 3) is a novel siderophore cephalosporin62,63) developed by Shionogi & Co., Ltd., that demonstrates notable activity against all the critical bacteria indicated by WHO, including carbapenem-resistant A. baumannii, carbapenem-resistant P. aeruginosa, and CRE including ESBL, KPC, and NDM-1.64–73) Furthermore, cefiderocol exhibits broad-spectrum activity against other ESKAPE pathogens.72,74,75) The chemical structure of cefiderocol was rationally designed to penetrate the outer cell membrane of Gram-negative bacteria and overcome resistance mechanisms.76) Efficient penetration of cefiderocol is performed by trapping a ferric ion with the catechol moiety as a siderophore, then the agent is actively transported into bacterial cells by a Trojan horse strategy.77) Consequently, the agent is retained in high concentrations in the periplasmic space, where it inhibits cell wall synthesis. In addition to its broad range of antimicrobial activity, the stability of cefiderocol against β-lactamases, including ESBL, KPC, and NDM-1, has been confirmed.78–80) Cefiderocol has been evaluated in phase III clinical trials81) and was submitted in a New Drug Application to the FDA in December 2018 and in a Marketing Authorization Application to the European Medicines Agency in March 2019.
BOS-228 (LYS228) (Fig. 3) is a potentially best-in-class monobactam developed by Boston Pharmaceuticals (Licensed from Novartis AG). It is now being evaluated in phase II clinical trials with QIDP status for the treatment of complicated urinary tract infections (cUTIs)82) and cIAIs.83) Monobactams are intrinsically stable to MBL.84) However, aztreonam (Fig. 3), a first-generation monobactam, is degraded by serine β-lactamase (SBL), which is often co-expressed with MBL in clinically relevant Enterobacteriaceae.85) BOS-228 was designed to exhibit stability against MBL and SBL. Its structural design concept was derived from the fact that carumonam and BO-1158, which possess a 4-cis configured β-lactam ring, are more stable to SBL than aztreonam.86) Based on this fact, a SAR study of 4-cis configured β-lactams was conducted.87,88) The introduction of oxazolidinone to the side branch of the monobactam, and cyclopropanation on the carboxylic acid moiety, increased its stability against SBL and MBL89) and increased its antimicrobial activity for NDM-1, KPC, and most ESBL.90–92) BOS-228 inhibits the growth of Escherichia coli by binding to penicillin-binding protein 3, a mechanism similar to that of aztreonam.93)
5. Novel β-Lactam/β-Lactamase Inhibitors for MDR Gram-negative Bacteria
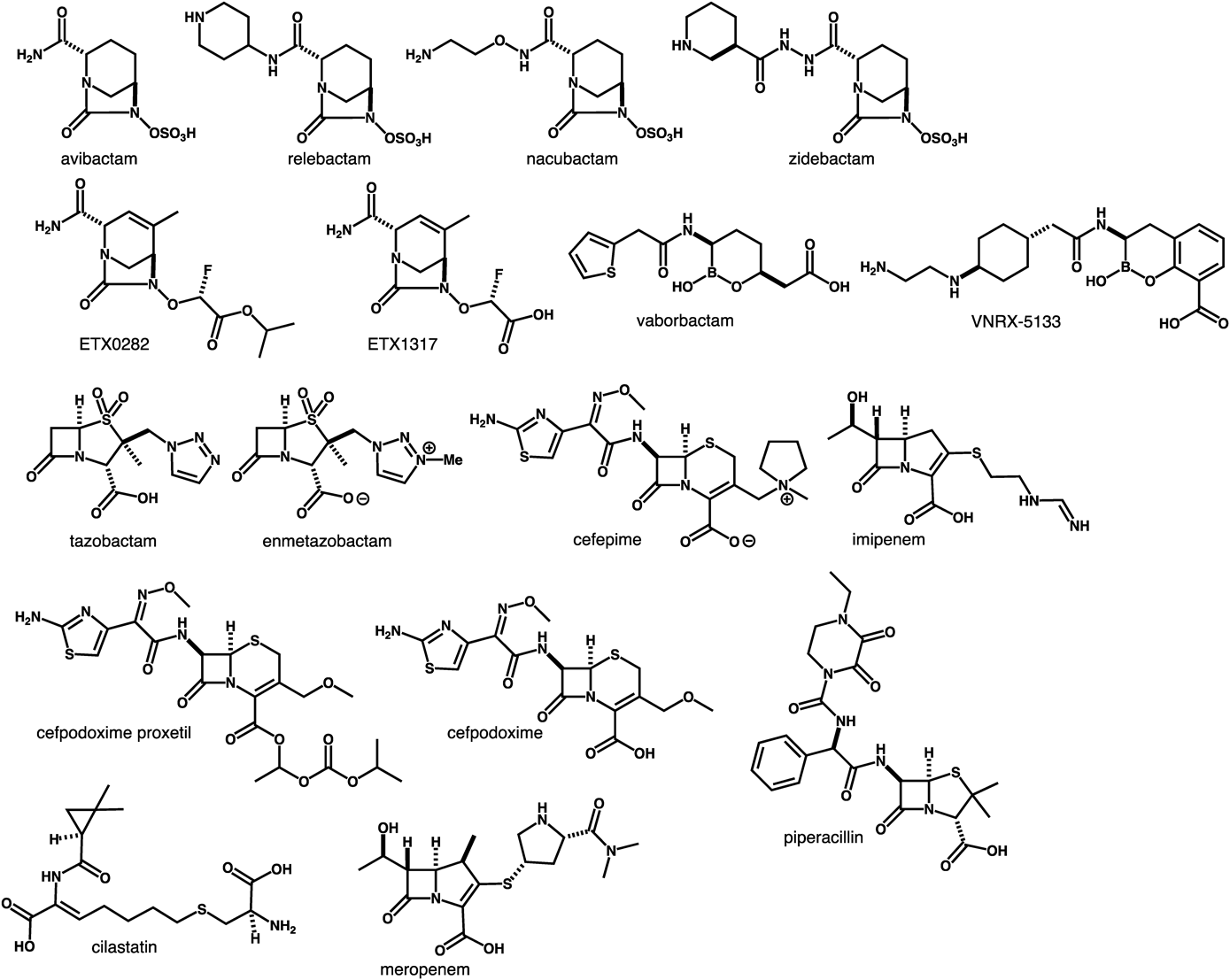
The combination of a β-lactam and a β-lactamase inhibitor is a well-established method for retaining the activity of β-lactam by suppressing the enzymatic hydrolysis of its β-lactam ring.94,95) However, several bacteria have already acquired resistance to β-lactam/β-lactamase inhibitor combinations. Recently, vaborbactam,96–98) a first-in-class boronic acid β-lactamase inhibitor that inhibits KPC-producing Enterobacteriaceae, was introduced to clinical practice. Vaborbactam/meropenem (Vabomere™) (Fig. 4) was approved for the treatment of cUTIs.99–104) The current clinical pipeline mainly contains structurally modified diazabicyclooctane-type β-lactamase inhibitors.105)
ETX0282CPDP is the first oral β-lactam/β-lactamase inhibitor combination drug consisting of cefpodoxime proxetil (oral cephalosporin) and ETX0282 (an oral prodrug of diazabicyclooctane-class β-lactamase inhibitor) (Fig. 4). ETX0282CPDP was developed by Entasis Therapeutics, Inc., and is being evaluated in phase I clinical trials for the treatment of infections caused by MDR Enterobacteriaceae, including CRE.106) Conversion of the ester moiety of ETX0282 in vivo produces ETX1317 (Fig. 4), which inhibits Ambler class A and C enzymes and select class D enzymes.107) Cefpodoxime proxetil,108) a third-generation semi-synthetic cephalosporin, is transformed to cefpodoxime (Fig. 4) as the active agent. In preclinical studies, ETX0282 restored the antimicrobial activity of cefpodoxime against Enterobacteriaceae that had acquired resistance to fluoroquinolones, cephalosporins, and carbapenems.107,109,110)
Imipenem/Cilastatin/Relebactam (MK-7655A). MK-7655A consists of three components: imipenem (carbapenem), cilastatin (renal DHP inhibitor), and relebactam (a novel β-lactamase inhibitor) (Fig. 4). It was developed by Merck & Co., Inc., and is being evaluated in phase III clinical trials with QIDP status and Fast Track designation.111,112) A combination of imipenem and cilastatin had been sold since 1987 under the brand name Primaxin.113) Relebactam, a diazabicyclooctane-class β-lactamase inhibitor containing a piperidine moiety, inhibits class A and class C β-lactamases114,115) and also restores the antimicrobial activity of imipenem against clinically relevant imipenem-resistant P. aeruginosa and K. pneumoniae.116–119) A SAR study of relebactam revealed that replacement of the piperidine with a pyrrolidine or azepane ring resulted in comparable β-lactamase inhibition activity and a synergistic effect with imipenem, whereas methylation of the amide moiety decreased the inhibition of KPC-2 and AmpC.120,121) FDA has accepted MK-7655A for review as a new drug application in the treatment of cUTIs122) and cIAIs caused by certain susceptible Gram-negative bacteria.
Nacubactam/Meropenem (OP0595/RG6080). Nacubactam (NAC, RG6080, OP0595)123–126) (Fig. 4) is a novel diazabicyclooctane-class β-lactamase inhibitor developed by NacuGen Therapeutics, Inc. (a joint venture of Meiji Seika Pharma Co., Ltd./Fedora Pharmaceuticals, Inc.). Nacubactam has received Fast Track and QIDP designations by the FDA, and is currently being evaluated in phase I clinical trials. Nacubactam inactivates class A and class C β-lactamases, and exhibits antibacterial activity against Enterobacteriaceae by binding to penicillin-binding protein 2.127) Furthermore, nacubactam enhances the activity of partnered β-lactams that bind to other penicillin-binding proteins.128) In non-clinical studies, the combination of nacubactam and meropenem129) (Fig. 4), a carbapenem antibiotic, was shown to have potent antimicrobial activity against cUTIs and severe respiratory tract infections caused by meropenem-resistant Enterobacteriaceae and CRE.130,131) Nacubactam/meropenem exhibits much better activity than that of piperacillin/tazobactam,123,132) as well as potent activity against carbapenem-resistant K. pneumoniae via a different mechanism from that of avibactam.133)
Cefepime/VNRX-5133 (Taniborbactam) (Fig. 4) is a combination of the fourth-generation cephalosporin cefepime134) and the next-generation β-lactamase inhibitor developed by VenatoRx Pharmaceuticals Inc. Its phase III trials in patients with cUTIs were initiated in August 2019. VNRX-5133, an injectable β-lactamase inhibitor containing a bicyclic boronate moiety, exhibits direct inhibitory activity against Ambler class A (ESBL, KPC), class B (NDM, VIM), class C (AmpC), and class D β-lactamases, and also enhances the activity of cefepime against CRE and carbapenem-resistant P. aeruginosa.135,136) VNRX-5133 was synthesized from 2-methoxy-3-methylbenzoic acid in 11 steps via stereoselective one-carbon homologation.137) Cefepime/VNRX-5133 exhibits potency against β-lactamase—producing Enterobacteriaceae and P. aeruginosa, including strains resistant to ceftazidime/avibactam, ceftolozane/tazobactam,138) and cefepime, meropenem, and piperacillin/tazobactam non-susceptible isolates.139,140) Furthermore, VNRX-5133 restores the antimicrobial activity of cefepime in the presence of NDM-1—producing Enterobacteriaceae.141) VNRX-5133 has QIDP status and Fast Track designation for the treatment of cUTIs and cIAIs.
WCK 5222 is a combination of cefepime and the novel β-lactam enhancer zidebactam (Fig. 4) that has been developed by Wockhardt Ltd. It is being evaluated in phase I clinical trials with QIDP status.142,143) Zidebactam, a diazabicyclooctane-class non-β-lactam antibiotic bearing a hydrazide moiety, exhibits a dual mechanism of action and high affinity for binding to penicillin-binding protein 2, resulting in the inhibition of Ambler class A and C β-lactamases.144–147) Zidebactam also acts as a β-lactam enhancer when combined with cephalosporin.148) While cefepime has antimicrobial activity against A. baumannii,149) cefepime/zidebactam exhibits better antimicrobial activity against A. baumannii in vitro and in vivo than cefepime alone.144,150,151) Accordingly, WCK 5222 shows potency for Enterobacteriaceae and P. aeruginosa producing clinically relevant β-lactamases, including ESBL, KPC, AmpC, and MBL.147,152–155)
Cefepime/Enmetazobactam (Formerly AAI101) (Fig. 4) is a combination of cefepime and a novel β-lactamase inhibitor developed by Allecra that has entered phase III clinical trials for the treatment of cUTIs with QIDP status and Fast Track designation. Enmetazobactam is a novel penicillanic acid sulfone ESBL inhibitor that exhibits potent antimicrobial activity for Gram-negative pathogens, and restores the activity of cefepime against class A (ESBL).156,157) Enmetazobactam was designed to increase the cell wall penetration ability of tazobactam for Gram-negative bacteria. The methylation of tazobactam’s triazole moiety yielded enmetazobactam as a zwitterion that exhibits high permeability of the bacterial cell wall. Cefepime is also a zwitterion.158) Enmetazobactam’s mechanism of β-lactamase inhibition differs from that of tazobactam.159) Cefepime is stable in the presence of AmpC and OXA-48. Cefepime/enmetazobactam outperforms piperacillin/tazobactam, and is as potent as meropenem toward Enterobacteriaceae and the subset of ESBL-producing E. coli and K. pneumoniae isolates.159–164)
AIC499/Unknown β-Lactamase Inhibitor. AIC499 is a novel β-lactam developed by AiCuris. It is currently being evaluated in phase I clinical trials. The combination of AIC499 and an unknown β-lactamase inhibitor shows potent antimicrobial activity against MDR strains of P. aeruginosa, A. baumannii, and β-lactamases such as Ambler class A, class B, class C, and some class D enzymes.165,166) However, limited information on this system is currently available, and the structure of AIC499 is not currently disclosed.
6. Novel Aminoglycosides for MDR Gram-negative Bacteria
There are currently no aminoglycoside antibiotics that exhibit efficacy in the treatment of infections caused by NDM-1—producing CRE and/or 16S ribosomal RNA methyltransferase-producing bacteria. TS3112 (Fig. 5), a semi-synthetic aminoglycoside derived from apramycin,167) was developed by the Institute of Microbial Chemistry (BIKAKEN) and Meiji Seika Pharma Co., Ltd.168,169) Chemical modification of apramycin led to TS3112 via stereo-inversion of the hydroxyl group at C-5 and attachment of an (S)-3-amino-2-hydroxypropionic acid to the amino group at C-4.” TS3112 is the first aminoglycoside antibiotic to exhibit potency against NDM-1—producing CRE and 16S ribosomal RNA methyltransferase-producing bacteria. Furthermore, TS3112 possesses a unique antibacterial profile, being effective against P. aeruginosa and A. baumannii, as well as against Gram-positive bacteria including Methicillin-resistant Staphylococcus aureus.170) Hence, TS3112 is expected to evolve into a key next-generation drug for the treatment of severe infections caused by MDR Gram-negative bacteria that are difficult to treat with existing drugs.
7. Novel Antibiotics for MDR Gram-negative Bacteria
Murepavadin (Formerly POL7080) (Fig. 6) is a novel antibiotic with a new mechanism of action,171) developed by Polyphor AG. It is being evaluated in phase III clinical trials with QIDP status for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia.172,173) Murepavadin is a peptide mimetic antibiotic comprising a cyclic peptide structure with 16 amino acids. It was synthesized with reference to the structure of protegrin I. Murepavadin selectively works against P. aeruginosa, whereas it is inactive against most other Gram-negative and -positive bacteria. Murepavadin has a novel mechanism that targets the outer membrane proteins of bacteria, namely the lipopolysaccharide transport protein D.174,175)In vitro antimicrobial evaluation has demonstrated that murepavadin is effective against MDR and highly drug-resistant P. aeruginosa, including carbapenem-resistant and colistin-resistant organisms. Furthermore, it exhibits 4- to 8-fold more potent antibacterial activity than colistin and polymyxin-B.176,177) Because of murepavadin’s novel mode of action, cross-resistance with other antibiotics is unlikely to occur.178) Furthermore, the P. aeruginosa-specific action of murepavadin is likely to prevent the diffusion of resistance to other pathogenic bacteria.
8. Conclusion
In this review, we have discussed promising antibiotics active against highly dangerous MDR Gram-negative bacteria, particularly those effective against CRE, MDR P. aeruginosa, and MDR A. baumannii. Our survey of these promising antibiotics revealed that natural product chemistry and synthetic organic chemistry still play key roles in the discovery and development of antibiotics. Innovative fluorocyclines have been developed by a synthetic chemical approach and by the SAR study of fully synthetic tetracycline scaffolds. Novel β-lactam antibiotics have been produced by chemical modification and the incorporation of functional units to the β-lactam skeleton. Simple strategic chemical transformation of the diazabicyclooctane framework has yielded several novel β-lactamase inhibitors, whereas the development of next-generation polymyxins and aminoglycosides have been achieved by a semi-synthetic method based on natural products. A synthetic biomimetic approach to a protegrin 1 mimic led to the discovery of murepavadin, which exhibits a novel mode of action. Thus, we believe that the fusion of natural product chemistry and modern synthetic chemistry will accelerate the discovery of next-generation antibiotics that exhibit unprecedented mechanisms.
Acknowledgments
I am grateful to Dr. Yoshiaki Takahashi, Dr. Eijiro Umemura and Dr. Masayuki Igarashi at the Institute of Microbial Chemistry (BIKAKEN) for their useful advice.
Conflict of Interest
The author declares no conflict of interest.
References
- 1) Pop-Vicas A., Opal S. M., Virulence, 5, 206–212 (2014).
- 2) Martens E., Demain A. L., J. Antibiot., 70, 520–526 (2017).
- 3) Exner M., Bhattacharya S., Christiansen B., Gebel J., Goroncy-Bermes P., Hartemann P., Heeg P., Ilschner C., Kramer A., Larson E., Merkens W., Mielke M., Oltmanns P., Ross B., Rotter M., Schmithausen R. M., Sonntag H.-G., Trautmann M., GMS Hyg. Infect. Control, 12, Doc05 (2017).
- 4) Gupta N., Limbago B. M., Patel J. B., Kallen A. J., Clin. Infect. Dis., 53, 60–67 (2011).
- 5) CDC, “Antibiotic resistance threats in the United States, 2013.”: ‹https://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf›, 2013.
- 6) WHO, “Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics.”: ‹http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/›, 2017.
- 7) Ramadan R. A., Gebriel M. G., Kadry H. M., Mosallem A., Infect. Drug Resist., 11, 1261–1269 (2018).
- 8) Rupp M. E., Fey P. D., Drugs, 63, 353–365 (2003).
- 9) Rodríguez-Baño J., Gutiérrez-Gutiérrez B., Machuca I., Pascual A., Clin. Microbiol. Rev., 31, e00079–e00017 (2018).
- 10) Tzouvelekis L. S., Markogiannakis A., Piperaki E., Souli M., Daikos G. L., Clin. Microbiol. Infect., 20, 862–872 (2014).
- 11) Fischbach M. A., Walsh C. T., Science, 325, 1089–1093 (2009).
- 12) McKenna M., Nature (London), 499, 394–396 (2013).
- 13) Bialvaei A. Z., Samadi Kafil H., Curr. Med. Res. Opin., 31, 707–721 (2015).
- 14) WHO, “Antibacterial Agents in Clinical Development: An analysis of the antibacterial clinical development pipeline, including tuberculosis.”: ‹http://www.who.int/medicines/areas/rational_use/antibacterial_agents_clinical_development/en/›, 2017.
- 15) The Pew Charitable Trusts, “Antibiotics Currently in Global Clinical Development.”: ‹https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development›, 2019.
- 16) Butler M. S., Blaskovich M. A. T., Cooper M. A., J. Antibiot., 70, 3–24 (2016).
- 17) Fernandes P., Martens E., Biochem. Pharmacol., 133, 152–163 (2017).
- 18) Taneja N., Kaur H., Microbiol. Insights, 9, 9–19 (2016).
- 19) Jones J. A., Virga K. G., Gumina G., Hevener K. E., Med. Chem. Commun., 7, 1694–1715 (2016).
- 20) Lakemeyer M., Zhao W., Mandl F. A., Hammann P., Sieber S. A., Angew. Chem. Int. Ed., 57, 14440–14475 (2018).
- 21) Liu F., Myers A. G., Curr. Opin. Chem. Biol., 32, 48–57 (2016).
- 22) Wright P. M., Seiple I. B., Myers A. G., Angew. Chem. Int. Ed., 53, 8840–8869 (2014).
- 23) Clark R. B., Hunt D. K., He M., Achorn C., Chen C.-L., Deng Y., Fyfe C., Grossman T. H., Hogan P. C., O’Brien W. J., Plamondon L., Rönn M., Sutcliffe J. A., Zhu Z., Xiao X.-Y., J. Med. Chem., 55, 606–622 (2012).
- 24) Xiao X.-Y., Hunt D. K., Zhou J., Clark R. B., Dunwoody N., Fyfe C., Grossman T. H., O’Brien W. J., Plamondon L., Rönn M., Sun C., Zhang W.-Y., Sutcliffe J. A., J. Med. Chem., 55, 597–605 (2012).
- 25) Zhanel G. G., Cheung D., Adam H., Zelenitsky S., Golden A., Schweizer F., Gorityala B., Lagace-Wiens P. R., Walkty A., Gin A. S., Hoban D. J., Karlowsky J. A., Drugs, 76, 567–588 (2016).
- 26) Zhou J., Xiao X.-Y., Plamondon L., Hunt D. K., Clark R. B., Zahler R. B., WO/2010/017470 A1 (2010).
- 27) Sutcliffe J. A., WO/2012/021829 A1 (2012).
- 28) Ronn M., Zhu Z., Hogan P. C., Zhang W.-Y., Niu J., Katz C. E., Dunwoody N., Gilicky O., Deng Y., Hunt D. K., He M., Chen C.-L., Sun C., Clark R. B., Xiao X.-Y., Org. Process Res. Dev., 17, 838–845 (2013).
- 29) Connors K. P., Housman S. T., Pope J. S., Russomanno J., Salerno E., Shore E., Redican S., Nicolau D. P., Antimicrob. Agents Chemother., 58, 2113–2118 (2014).
- 30) Solomkin J. S., Ramesh M. K., Cesnauskas G., Novikovs N., Stefanova P., Sutcliffe J. A., Walpole S. M., Horn P. T., Antimicrob. Agents Chemother., 58, 1847–1854 (2014).
- 31) Bassetti M., Righi E., Expert Opin. Investig. Drugs, 23, 1575–1584 (2014).
- 32) Grossman T. H., Starosta A. L., Fyfe C., O’Brien W., Rothstein D. M., Mikolajka A., Wilson D. N., Sutcliffe J. A., Antimicrob. Agents Chemother., 56, 2559–2564 (2012).
- 33) Grossman T. H., Pillar C. M., Sahm D. F., Sutcliffe J. A., Antimicrob. Agents Chemother., 59, 2426–2427 (2015).
- 34) Abdallah M., Olafisoye O., Cortes C., Urban C., Landman D., Quale J., Antimicrob. Agents Chemother., 59, 1802–1805 (2015).
- 35) Sutcliffe J. A., O’Brien W., Fyfe C., Grossman T. H., Antimicrob. Agents Chemother., 57, 5548–5558 (2013).
- 36) Zhang Y., Lin X., Bush K., J. Antibiot., 69, 600–604 (2016).
- 37) Livermore D. M., Mushtaq S., Warner M., Woodford N., Antimicrob. Agents Chemother., 60, 3840–3844 (2016).
- 38) Peterson L. R., Int. J. Antimicrob. Agents, 32 (Suppl 4), S215–S222 (2008).
- 39) Monogue M. L., Thabit A. K., Hamada Y., Nicolau D. P., Antimicrob. Agents Chemother., 60, 5001–5005 (2016).
- 40) Thabit A. K., Monogue M. L., Nicolau D. P., Antimicrob. Agents Chemother., 60, 5072–5075 (2016).
- 41) Zhao M., Lepak A. J., Marchillo K., VanHecker J., Andes D. R., Antimicrob. Agents Chemother., 61, e00250–e00217 (2017).
- 42) Xiao X.-Y., Clark R. B., Hunt D. K., Sun C., Ronn M., Zhang W.-Y., He M., WO/2014/036502 A2 (2014).
- 43) Xiao X.-Y., Dumas J. P., Hunt D. K., Sun C., Zhao P., WO/2018/045084 A1 (2018).
- 44) Sun C., Deng Y., Hunt D., Fyfe C., Kerstein K., Xiao X., Microbe A. S. M., 2017 Sunday-332, New Orleans, LA, U.S.A. (2017).
- 45) Falagas M. E., Skalidis T., Vardakas K. Z., Voulgaris G. L., Papanikolaou G., Legakis N., Int. J. Antimicrob. Agents, 52, 269–271 (2018).
- 46) Fyfe C., Barry R., Close B., Kerstein K., Nordmann P., Newman J., Microbe A. S. M., 2018 Poster-655, Atlanta, GA, U.S.A., 2018.
- 47) Lenhard J. R., Bulman Z. P., Tsuji B. T., Kaye K. S., Antibiotics, 8, 42 (2019).
- 48) Rice L. B., J. Infect. Dis., 197, 1079–1081 (2008).
- 49) Arends S. J. R., Rhomberg P. R., Lister T., Cotroneo N., Rubio A., Flamm R. K., Mendes R. E., ASM-ESCMID 2018 Poster #80 Lisbon, Portugal, 2018.
- 50) Arends S. J. R., Rhomberg P. R., Lister T., Cotroneo N., Rubio A., Flamm R. K., Mendes R. E., ASM-ESCMID 2018 Poster #81 Lisbon, Portugal (2018).
- 51) Akhoundsadegh N., Belanger C. R., Hancock R. E. W., Antimicrob. Agents Chemother., 63, e00935–e00919 (2019).
- 52) Brown P., Boakes S., Duperchy E., Abdulle O., Rivers D., Simonovic M., Singh J., Coleman S., Dawson M. J., ASM Microbe 2019 Poster-793 San Francisco, CA, U.S.A., 2019.
- 53) Brown P., Boakes S., Duperchy E., Rivers D., Singh J., Dawson M. J., ASM ESCMID 2018 Poster-145 Lisbon, Portugal, 2018.
- 54) Brown P., Abbott E., Abdulle O., Boakes S., Coleman S., Divall N., Duperchy E., Moss S., Rivers D., Simonovic M., Singh J., Stanway S., Wilson A., Dawson M. J., ACS Infect. Dis., 5, 1645–1656 (2019).
- 55) Mingeot-Leclercq M.-P., Tulkens P. M., Denamur S., Vaara T., Vaara M., Peptides, 35, 248–252 (2012).
- 56) Vaara M., J. Antimicrob. Chemother., 68, 1213–1219 (2013).
- 57) Lister T., WO/2017/197291 A1 (2017).
- 58) Coleman S., Shastri P. N., WO/2017/197390 A1 (2017).
- 59) Vaara M., Siikanen O., Apajalahti J., Fox J., Frimodt-Møller N., He H., Poudyal A., Li J., Nation R. L., Vaara T., Antimicrob. Agents Chemother., 54, 3341–3346 (2010).
- 60) Corbett D., Wise A., Langley T., Skinner K., Trimby E., Birchall S., Dorali A., Sandiford S., Williams J., Warn P., Vaara M., Lister T., Antimicrob. Agents Chemother., 61, e00200–e00217 (2017).
- 61) Zurawski D. V., Reinhart A. A., Alamneh Y. A., Pucci M. J., Si Y., Abu-Taleb R., Shearer J. P., Demons S. T., Tyner S. D., Lister T., Antimicrob. Agents Chemother., 61, e01239–e01217 (2017).
- 62) Kawasaki H., Kojima N., Fujihira A., Takahashi K., Matsubara F., Matsuoka N., WO/2016/035846 A1 (2016).
- 63) Matsubara F., Kurita T., Nagamatsu D., WO/2016/035845 A1 (2016).
- 64) Ito A., Kohira N., Bouchillon S. K., West J., Rittenhouse S., Sader H. S., Rhomberg P. R., Jones R. N., Yoshizawa H., Nakamura R., Tsuji M., Yamano Y., J. Antimicrob. Chemother., 71, 670–677 (2016).
- 65) Kohira N., West J., Ito A., Ito-Horiyama T., Nakamura R., Sato T., Rittenhouse S., Tsuji M., Yamano Y., Antimicrob. Agents Chemother., 60, 729–734 (2016).
- 66) Karlowsky J. A., Hackel M. A., Tsuji M., Yamano Y., Echols R., Sahm D. F., Int. J. Antimicrob. Agents, 53, 456–466 (2019).
- 67) Matsumoto S., Singley C. M., Hoover J., Nakamura R., Echols R., Rittenhouse S., Tsuji M., Yamano Y., Antimicrob. Agents Chemother., 61, e00700–e00717 (2017).
- 68) Hackel M. A., Tsuji M., Yamano Y., Echols R., Karlowsky J. A., Sahm D. F., Antimicrob. Agents Chemother., 61, e00093–e00017 (2017).
- 69) Hackel M. A., Tsuji M., Yamano Y., Echols R., Karlowsky J. A., Sahm D. F., Antimicrob. Agents Chemother., 62, e01968–e01917 (2018).
- 70) Zhanel G. G., Golden A. R., Zelenitsky S., Wiebe K., Lawrence C. K., Adam H. J., Idowu T., Domalaon R., Schweizer F., Zhanel M. A., Lagacé-Wiens P. R. S., Walkty A. J., Noreddin A., Lynch J. P. III, Karlowsky J. A., Drugs, 79, 271–289 (2019).
- 71) Jacobs M. R., Abdelhamed A. M., Good C. E., Rhoads D. D., Hujer K. M., Hujer A. M., Domitrovic T. N., Rudin S. D., Richter S. S., van Duin D., Kreiswirth B. N., Greco C., Fouts D. E., Bonomo R. A., Antimicrob. Agents Chemother., 63, e01801–e01818 (2019).
- 72) Falagas M. E., Skalidis T., Vardakas K. Z., Legakis N. J., J. Antimicrob. Chemother., 72, 1704–1708 (2017).
- 73) Monogue M. L., Tsuji M., Yamano Y., Echols R., Nicolau D. P., Antimicrob. Agents Chemother., 61, e01022–e01017 (2017).
- 74) Kazmierczak K. M., Tsuji M., Wise M. G., Hackel M., Yamano Y., Echols R., Sahm D. F., Int. J. Antimicrob. Agents, 53, 177–184 (2019).
- 75) Ito A., Sato T., Ota M., Takemura M., Nishikawa T., Toba S., Kohira N., Miyagawa S., Ishibashi N., Matsumoto S., Nakamura R., Tsuji M., Yamano Y., Antimicrob. Agents Chemother., 62, e01454–e01417 (2018).
- 76) Aoki T., Yoshizawa H., Yamawaki K., Yokoo K., Sato J., Hisakawa S., Hasegawa Y., Kusano H., Sano M., Sugimoto H., Nishitani Y., Sato T., Tsuji M., Nakamura R., Nishikawa T., Yamano Y., Eur. J. Med. Chem., 155, 847–868 (2018).
- 77) Ito A., Nishikawa T., Matsumoto S., Yoshizawa H., Sato T., Nakamura R., Tsuji M., Yamano Y., Antimicrob. Agents Chemother., 60, 7396–7401 (2016).
- 78) Ito-Horiyama T., Ishii Y., Ito A., Sato T., Nakamura R., Fukuhara N., Tsuji M., Yamano Y., Yamaguchi K., Tateda K., Antimicrob. Agents Chemother., 60, 4384–4386 (2016).
- 79) Ito A., Nishikawa T., Ota M., Ito-Horiyama T., Ishibashi N., Sato T., Tsuji M., Yamano Y., J. Antimicrob. Chemother., 73, 3049–3052 (2018).
- 80) Poirel L., Kieffer N., Nordmann P., Int. J. Antimicrob. Agents, 52, 866–867 (2018).
- 81) Portsmouth S., van Veenhuyzen D., Echols R., Machida M., Ferreira J. C. A., Ariyasu M., Tenke P., Nagata T. D., Lancet Infect. Dis., 18, 1319–1328 (2018).
- 82) Reck F., WO/2019/092180 A1 (2019).
- 83) Reck F., WO/2019/058346 A1 (2019).
- 84) Felici A., Amicosante G., Antimicrob. Agents Chemother., 39, 192–199 (1995).
- 85) Biedenbach D. J., Kazmierczak K., Bouchillon S. K., Sahm D. F., Bradford P. A., Antimicrob. Agents Chemother., 59, 4239–4248 (2015).
- 86) Matsuda K., Nakagawa S., Nakano F., Inoue M., Mitsuhashi S., J. Antimicrob. Chemother., 19, 753–760 (1987).
- 87) Aulakh V. S., Casarez A., Lin X., Lindvall M., McEnroe G., Moser H. E., Reck F., Tjandra M., Simmons R. L., Yifru A., Zhu Q., U.S. 2015/0266867 A1 (2015).
- 88) Aubin E., Casarez A., Fisch A., Li Z., Lindvall M., Moser H. E., Mutz M., Reck F., Riebesehl B. U., Schoenhentz M., Sethuraman V., Simmons R. L., WO/2017/050218 (2017).
- 89) Reck F., Bermingham A., Blais J., et al., Bioorg. Med. Chem. Lett., 28, 748–755 (2018).
- 90) Blais J., Lopez S., Li C., Ruzin A., Ranjitkar S., Dean C. R., Leeds J. A., Casarez A., Simmons R. L., Reck F., Antimicrob. Agents Chemother., 62, e00552–e00518 (2018).
- 91) Weiss W. J., Pulse M. E., Nguyen P., Growcott E. J., Antimicrob. Agents Chemother., 63, e02214–e02218 (2019).
- 92) Jones A. K., Ranjitkar S., Lopez S., Li C., Blais J., Reck F., Dean C. R., Antimicrob. Agents Chemother., 62, e01202–e01218 (2018).
- 93) Dean C. R., Barkan D. T., Bermingham A., et al., Antimicrob. Agents Chemother., 62, e01200–e01218 (2018).
- 94) Drawz S. M., Bonomo R. A., Clin. Microbiol. Rev., 23, 160–201 (2010).
- 95) Bush K., Bradford P. A., Cold Spring Harb. Perspect. Med., 6, a025247 (2016).
- 96) Lomovskaya O., Sun D., Rubio-Aparicio D., Nelson K., Tsivkovski R., Griffith D. C., Dudley M. N., Antimicrob. Agents Chemother., 61, e01443–e01417 (2017).
- 97) Hecker S. J., Reddy K. R., Totrov M., et al., J. Med. Chem., 58, 3682–3692 (2015).
- 98) Langley G. W., Cain R., Tyrrell J. M., Hinchliffe P., Calvopiña K., Tooke C. L., Widlake E., Dowson C. G., Spencer J., Walsh T. R., Schofield C. J., Brem J., Bioorg. Med. Chem. Lett., 29, 1981–1984 (2019).
- 99) Dhillon S., Drugs, 78, 1259–1270 (2018).
- 100) Castanheira M., Rhomberg P. R., Flamm R. K., Jones R. N., Antimicrob. Agents Chemother., 60, 5454–5458 (2016).
- 101) Zhanel G. G., Lawrence C. K., Adam H., Schweizer F., Zelenitsky S., Zhanel M., Lagacé-Wiens P. R. S., Walkty A., Denisuik A., Golden A., Gin A. S., Hoban D. J., Lynch J. P. III, Karlowsky J. A., Drugs, 78, 65–98 (2018).
- 102) Zhou M., Yang Q., Lomovskaya O., Sun D., Kudinha T., Xu Z., Zhang G., Chen X., Xu Y., J. Antimicrob. Chemother., 73, 2789–2796 (2018).
- 103) Bassetti M., Giacobbe D. R., Patel N., Tillotson G., Massey J., Adv. Ther., 36, 1771–1777 (2019).
- 104) Jorgensen S. C. J., McDonald P., Mynatt R. P., Pogue J. M., Lerner S. A., Dhar S., Salimnia H., Rybak M. J., J. Antimicrob. Chemother., 73, 3529–3531 (2018).
- 105) Coleman K., Curr. Opin. Microbiol., 14, 550–555 (2011).
- 106) Comita-Prevoir J., Durand-Reville T. F., Gauthier L., O’donnell J., Romero J., Tommasi R., Verheijen J., Cunera, Wu F., Wu X., Zhang J., Basarab G. S., Moss B., WO/2018/053215 A1 (2018).
- 107) McLeod S., Carter N., Moussa S., Mueller J., Tommasi R., Miller A., Microbe A. S. M., 2019 Sunday-AAR-714 San Francisco, CA, U.S.A. (2019).
- 108) Todd W. M., Int. J. Antimicrob. Agents, 4, 37–62 (1994).
- 109) O’Donnell J., Chen A., Tanudra A., Mueller J., Tommasi R., Miller A., Carter N., McLeod S., Comita-Prevoir J., Durand-Réville T., Microbe A. S. M., 2017 Session-198 Saturday-278, New Orleans, LA, U.S.A. (2017).
- 110) McLeod S., Carter N., Hackel M., Badal R., Mueller J., Tommasi R., Miller A., Microbe A. S. M., 2018 Friday-603 Atlanta, GA U.S.A. (2018).
- 111) Rizk M. L., Rhee E. G., Jumes P. A., Gotfried M. H., Zhao T., Mangin E., Bi S., Chavez-Eng C. M., Zhang Z., Butterton J. R., Antimicrob. Agents Chemother., 62, e01411–e01417 (2018).
- 112) Lucasti C., Vasile L., Sandesc D., Venskutonis D., McLeroth P., Lala M., Rizk M. L., Brown M. L., Losada M. C., Pedley A., Kartsonis N. A., Paschke A., Antimicrob. Agents Chemother., 60, 6234–6243 (2016).
- 113) Lyon J. A., Drug Intell. Clin. Pharm., 19, 895–899 (1985).
- 114) Livermore D. M., Jamrozy D., Mushtaq S., Nichols W. W., Young K., Woodford N., J. Antimicrob. Chemother., 72, 3342–3348 (2017).
- 115) Tooke C. L., Hinchliffe P., Lang P. A., Mulholland A. J., Brem J., Schofield C. J., Spencer J., Antimicrob. Agents Chemother., 63, e00564–e00519 (2019).
- 116) Lob S. H., Hackel M. A., Kazmierczak K. M., Young K., Motyl M. R., Karlowsky J. A., Sahm D. F., Antimicrob. Agents Chemother., 61, e02209–e02216 (2017).
- 117) Karlowsky J. A., Lob S. H., Kazmierczak K. M., Hawser S. P., Magnet S., Young K., Motyl M. R., Sahm D. F., J. Antimicrob. Chemother., 73, 1872–1879 (2018).
- 118) Powles M. A., Galgoci A., Misura A., Colwell L., Dingley K. H., Tang W., Wu J., Blizzard T., Motyl M., Young K., Antimicrob. Agents Chemother., 62, e02577–e02517 (2018).
- 119) Wu J., Racine F., Wismer M. K., Young K., Carr D. M., Xiao J. C., Katwaru R., Si Q., Harradine P., Motyl M., Bhagunde P. R., Rizk M. L., Antimicrob. Agents Chemother., 62, e02323–e02317 (2018).
- 120) Blizzard T. A., Chen H., Kim S., et al., Bioorg. Med. Chem. Lett., 24, 780–785 (2014).
- 121) Liu Z., Yasuda N., Simeone M., Reamer R. A., J. Org. Chem., 79, 11792–11796 (2014).
- 122) Sims M., Mariyanovski V., McLeroth P., Akers W., Lee Y.-C., Brown M. L., Du J., Pedley A., Kartsonis N. A., Paschke A., J. Antimicrob. Chemother., 72, 2616–2626 (2017).
- 123) Morinaka A., Tsutsumi Y., Yamada M., Suzuki K., Watanabe T., Abe T., Furuuchi T., Inamura S., Sakamaki Y., Mitsuhashi N., Ida T., Livermore D. M., J. Antimicrob. Chemother., 70, 2779–2786 (2015).
- 124) Livermore D. M., Warner M., Mushtaq S., Woodford N., Antimicrob. Agents Chemother., 60, 554–560 (2016).
- 125) Tadiparthi R., Patil V. J., Dixit P., Patel M. V., WO/2016/120752 A1 (2016).
- 126) Bhagwat S., Patel M. V., WO/2016/151543 A1 (2016).
- 127) Livermore D. M., Mushtaq S., Warner M., Woodford N., J. Antimicrob. Chemother., 70, 3032–3041 (2015).
- 128) Mushtaq S., Vickers A., Woodford N., Haldimann A., Livermore D. M., J. Antimicrob. Chemother., 74, 953–960 (2019).
- 129) Baldwin C. M., Lyseng-Williamson K. A., Keam S. J., Drugs, 68, 803–838 (2008).
- 130) Monogue M. L., Giovagnoli S., Bissantz C., Zampaloni C., Nicolau D. P., Antimicrob. Agents Chemother., 62, e02596–e02517 (2018).
- 131) Asempa T. E., Motos A., Abdelraouf K., Bissantz C., Zampaloni C., Nicolau D. P., Antimicrob. Agents Chemother., 63, e02382–e02318 (2019).
- 132) Bryson H. M., Brogden R. N., Drugs, 47, 506–535 (1994).
- 133) Barnes M. D., Taracila M. A., Good C. E., Bajaksouzian S., Rojas L. J., van Duin D., Kreiswirth B. N., Jacobs M. R., Haldimann A., Papp-Wallace K. M., Bonomo R. A., Antimicrob. Agents Chemother., 63, e00432–e00419 (2019).
- 134) Barradell L. B., Bryson H. M., Drugs, 47, 471–505 (1994).
- 135) Burns C., J., Daigle D., Liu B., Mcgarry D., Pevear D. C., Trout R. E. L., WO/2014/089365 A1 (2014).
- 136) Burns C. J., Pevear D. C., Xerri L., Henkel T., Mcgarry D., Rosen L., Brenner G., Arlin J.-B., Fernandez Casares A., WO/2018/165048 A1 (2018).
- 137) Krajnc A., Brem J., Hinchliffe P., Calvopiña K., Panduwawala T. D., Lang P. A., Kamps J. J. A. G., Tyrrell J. M., Widlake E., Saward B. G., Walsh T. R., Spencer J., Schofield C. J., J. Med. Chem., 62, 8544–8556 (2019).
- 138) Daigle D., Hamrick J., Chatwin C., Kurepina N., Kreiswirth B. N., Shields R. K., Oliver A., Clancy C. J., Nguyen M.-H., Pevear D., Xerri L., Open Forum Infect. Dis., 5 (suppl. 1), S419–S420 (2018).
- 139) Hackel M., Sahm D., ECCMID 2019 Poster-1175 Amsterdam, Netherlands (2019).
- 140) Hackel M., Sahm D., Microbe A. S. M., 2019 Sunday-AAR-772 San Francisco, CA, U.S.A. (2019).
- 141) Piccirilli A., Segatore B., Daigle D., Amicosante G., Perilli M., ECCMID 2019 Poster-1178 Amsterdam, Netherlands (2019).
- 142) Preston R. A., Mamikonyan G., DeGraff S., Chiou J., Kemper C. J., Xu A., Mastim M., Yeole R., Chavan R., Patel A., Friedland H. D., Bhatia A., Antimicrob. Agents Chemother., 63, e01484–e01418 (2019).
- 143) Rodvold K. A., Gotfried M. H., Chugh R., Gupta M., Patel A., Chavan R., Yeole R., Friedland H. D., Bhatia A., Antimicrob. Agents Chemother., 62, e00682–e00618 (2018).
- 144) Sader H. S., Rhomberg P. R., Flamm R. K., Jones R. N., Castanheira M., J. Antimicrob. Chemother., 72, 1696–1703 (2017).
- 145) Livermore D. M., Mushtaq S., Warner M., Vickers A., Woodford N., J. Antimicrob. Chemother., 72, 1373–1385 (2017).
- 146) Moya B., Barcelo I. M., Cabot G., Torrens G., Palwe S., Joshi P., Umarkar K., Takalkar S., Periasamy H., Bhagwat S., Patel M., Bou G., Oliver A., Antimicrob. Agents Chemother., 63, e00128–e00119 (2019).
- 147) Papp-Wallace K. M., Nguyen N. Q., Jacobs M. R., Bethel C. R., Barnes M. D., Kumar V., Bajaksouzian S., Rudin S. D., Rather P. N., Bhavsar S., Ravikumar T., Deshpande P. K., Patil V., Yeole R., Bhagwat S. S., Patel M. V., van den Akker F., Bonomo R. A., J. Med. Chem., 61, 4067–4086 (2018).
- 148) Bhagwat S. S., Periasamy H., Takalkar S. S., Palwe S. R., Khande H. N., Patel M. V., Antimicrob. Agents Chemother., 63, e02146–e02118 (2019).
- 149) Moya B., Barcelo I. M., Bhagwat S., Patel M., Bou G., Papp-Wallace K. M., Bonomo R. A., Oliver A., Antimicrob. Agents Chemother., 61, e01238–e01217 (2017).
- 150) Avery L. M., Abdelraouf K., Nicolau D. P., Antimicrob. Agents Chemother., 62, e00948–e00918 (2018).
- 151) Almarzoky Abuhussain S. S., Avery L. M., Abdelraouf K., Nicolau D. P., Antimicrob. Agents Chemother., 63, e01931–e01918 (2019).
- 152) Thomson K. S., AbdelGhani S., Snyder J. W., Thomson G. K., Antibiotics, 8, 32 (2019).
- 153) Sader H. S., Castanheira M., Huband M., Jones R. N., Flamm R. K., Antimicrob. Agents Chemother., 61, e00072–e00017 (2017).
- 154) Khan Z., Iregui A., Landman D., Quale J., J. Antimicrob. Chemother., 74, 2938–2942 (2019).
- 155) Monogue M. L., Tabor-Rennie J., Abdelraouf K., Nicolau D. P., Antimicrob. Agents Chemother., 63, e00233–e00219 (2019).
- 156) Faini A., Forzatti M., Fogliato G., Biondi S., WO/2015/173378 A2 (2015).
- 157) Lamonica A., Forzatti M., Biondi S., WO/2015/067787 A1 (2015).
- 158) Endimiani A., Perez F., Bonomo R. A., Expert Rev. Anti Infect. Ther., 6, 805–824 (2008).
- 159) Papp-Wallace K. M., Bethel C. R., Caillon J., Barnes M. D., Potel G., Bajaksouzian S., Rutter J. D., Reghal A., Shapiro S., Taracila M. A., Jacobs M. R., Bonomo R. A., Jacqueline C., Antimicrob. Agents Chemother., 63, e00105–e00119 (2019).
- 160) Morrissey I., Magnet S., Hawser S., Shapiro S., Knechtle P., Antimicrob. Agents Chemother., 63, e00514–e00519 (2019).
- 161) Papp-Wallace K. M., Bethel C. R., Barnes M. D., Rutter J. D., Taracila M. A., Bajaksouzian S., Jacobs M. R., Bonomo R. A., Open Forum Infect. Dis., 4 (Suppl. 1), S375 (2017).
- 162) Crandon J. L., Nicolau D. P., Pathogens, 4, 620–625 (2015).
- 163) Crandon J. L., Nicolau D. P., Antimicrob. Agents Chemother., 59, 2688–2694 (2015).
- 164) Belley A., Huband M. D., Fedler K. A., Watters A. A., Flamm R. K., Shapiro S., Knechtle P., J. Clin. Microbiol., 57, e00607–e00619 (2019).
- 165) Isler B., Doi Y., Bonomo R. A., Paterson D. L., Antimicrob. Agents Chemother., 63, e01110–e01118 (2019).
- 166) Kostyanev T., Bonten M. J. M., O’Brien S., Steel H., Ross S., François B., Tacconelli E., Winterhalter M., Stavenger R. A., Karlén A., Harbarth S., Hackett J., Jafri H. S., Vuong C., MacGowan A., Witschi A., Angyalosi G., Elborn J. S., deWinter R., Goossens H., J. Antimicrob. Chemother., 71, 290–295 (2016).
- 167) O’Connor S., Lam L. K. T., Jones N. D., Chaney M. O., J. Org. Chem., 41, 2087–2092 (1976).
- 168) Takahashi Y., Igarashi M., J. Antibiot., 71, 4–14 (2018).
- 169) Takahashi Y., Umemura E., Ida T., Igarashi M., WO/2017/018528 (2017).
- 170) Umemura E., Takahashi Y., Igarashi M., Hayashi C., Shibasaki M., Yamada K., Ida T., Yonezawa M., Ago K., Microbe A. S. M., 2017, Poster Sunday-335 New Orleans, LA, U.S.A. (2017).
- 171) Martin-Loeches I., Dale G. E., Torres A., Expert Rev. Anti Infect. Ther., 16, 259–268 (2018).
- 172) Wach A., Dembowsky K., Dale G. E., Antimicrob. Agents Chemother., 62, e02355–e02317 (2018).
- 173) Dale G. E., Halabi A., Petersen-Sylla M., Wach A., Zwingelstein C., Antimicrob. Agents Chemother., 62, e00490–e00418 (2018).
- 174) Srinivas N., Jetter P., Ueberbacher B. J., et al., Science, 327, 1010–1013 (2010).
- 175) Werneburg M., Zerbe K., Juhas M., Bigler L., Stalder U., Kaech A., Ziegler U., Obrecht D., Eberl L., Robinson J. A., ChemBioChem, 13, 1767–1775 (2012).
- 176) Sader H. S., Dale G. E., Rhomberg P. R., Flamm R. K., Antimicrob. Agents Chemother., 62, e00311–e00318 (2018).
- 177) Sader H. S., Flamm R. K., Dale G. E., Rhomberg P. R., Castanheira M., J. Antimicrob. Chemother., 73, 2400–2404 (2018).
- 178) Romano K. P., Warrier T., Poulsen B. E., Nguyen P. H., Loftis A. R., Saebi A., Pentelute B. L., Hung D. T., Antimicrob. Agents Chemother., 63, e00511–e00519 (2019).