Current Topics: Reviews
Antibody–Drug Conjugate Payloads; Study of Auristatin Derivatives
2020 Volume 68 Issue 3 Pages 201-211
Details
2020 Volume 68 Issue 3 Pages 201-211
Auristatins are important payloads used in antibody drug conjugates (ADCs), and the most well-known compound family member, monomethyl auristatin (MMAE), is used in two Food and Drug Administration (FDA)-approved ADCs, Adcetris® and Polivy®. Multiple other auristatin-based ADCs are currently being evaluated in human clinical trials and further studies on this class of molecule are underway by several academic and industrial research groups. Our group's main focus is to investigate the structure–activity relationships (SAR) of novel auristatins with the goal of applying these to next generation ADCs. Modifications of the auristatin backbone scaffold have been widely reported in the chemical literature focusing on the terminal subunits: P1 (N-terminus) and P5 (C-terminus). Our approach was to modulate the activity and hydrophilic character through modifications of the central subunits P2-P3-P4 and thorough SAR study on the P5 subunit. Novel hydrophilic auristatins were observed to have greater potency in vitro and displayed enhanced in vivo antitumor activity when conjugated via protease-cleavable linkers and delivered intracellularly. Analysis of ADC aggregation also indicated that novel hydrophilic payloads enabled the synthesis of high-drug-to-antibody ratio (DAR) ADCs that were resistant to aggregation. Modification of the central peptide subunits also resulted in auristatins with potent cytotoxic activity in vitro and these azide-modified auristatins contain a handle for linker attachment from the central portion of the auristatin backbone.
Antibody drug conjugates (ADCs) enable the delivery of cytotoxic payloads to specific antigen-expressing cancer cells via monoclonal antibodies to afford a class of targeted cancer treatment with clinically relevant therapeutic window.1) Typically, ADC payloads are restricted to highly potent cytotoxic molecules (pM to nM IC50) and rely on delivery of these active molecules to the targeted cells or tumor microenvironment.1,2) Many commonly used ADC payloads are derived from highly cytotoxic natural products and some of the most well-known ADC payloads belong to several classes based on mechanism of action: tubulin interactors3–5) (maytansinoids, auristatins), DNA modifiers3,6–8) (calicheamicin, duocarmycins, camptothecin), and RNA inhibitors9,10) (amatoxins).
To date, five ADCs such as gemtuzumab ozogamicin (Mylotarg®, 1), inotuzumab ozogamicin (CMC-544, Besponsa®, 2), ado-trastuzumab emtansine (T-DM1, Kadcyla®, 3), brentuximab vedotin (SGN-35, Adcetris®, 4), and polatuzumab vedotin (RG7596/DCDS-4501A, Polivy®, 5) have been approved.11–14) These commercial ADCs are described in Fig. 1 and contain payloads based on calicheamicin, maytansine, and auristatin E.

AcBut, 4-(4′-acetylphenoxy) butanoic acid; mc-VC-Paba (vc), maleimidocaproyl valine-citrulline p-aminobenzoyloxycarbamoyl; SMCC, maleimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate.
The Food and Drug Administration (FDA) approved auristatin-based ADCs, followed by Adcetris® (4),13) recently Polivy® (5)12) became 2nd approved ADC in 2019. Adcetris® (4), is an anti-CD30 ADC comprising a cleavable linker by proteases, such as cathepsin B and monomethyl auristatin (MMAE, 7d) as a payload with an average drug-to-antibody ratio (DAR) of 4. Polivy® (5) is an anti-CD79b ADC comprising the same linker and payload as Adcetris® (4) with DAR of 3.5. In addition, including our ADC, Enfortumab vedotin,15) over 10 ADCs with auristatins such as MMAE (7d) or monomethyl auristatin F (MMAF, 7e) as payload are currently in clinical trials (Table 1), which account for half of all ADCs in clinical trials at present.11,14) Over the last few decades, many research groups have studied auristatin analogues and its ADCs and this research area has been extensively reviewed both on academic and industrial sides.11,16) Thus auristatins are attractive toxic compounds on ADCs and we also have investigated the structure–activity relationship (SAR) and auristatins ADCs to create the next generation ADCs.17–20)
| ADC | Company | mAb mode | Target | Payload | Linker | Clinical phase | Indications |
|---|---|---|---|---|---|---|---|
| Adcetris®(brentuximab vedotin, SGN-35) | Seattle Genetics | Chi IgG1 | CD30 | MMAE | vc | Approved in 2011 | ALCL, HL |
| Polivy® (Polatuzumab vedotin, RG7596/DCDS-4501 A | Roche Genentech | Hz IgG1 | CD79b | MMAE | vc | Approved in 2019 | Follicular NHL and DLBCL |
| Enfortumab vedotin (AGS-22ME) | Astellas (Agensys) | IgG1 | Nectin 4 | MMAE | vc | Phase III (BLA Review) | Metastatic urothelial cancer |
| Telisotuzumab vedotin (ABBV-399) | AbbVie/Pierre Fabre | Engineered IgG1 | HGFR (cMet) | MMAE | vc | Phase II | NSCL |
| Tisotumab vedotin (HuMax-TF-ADC) | Genmab | Human IgG1 | Tissue factor (CD142) | MMAE | vc | Phase II | Multiple solid tumors |
| Anti-PSMA ADC | Progenics | IgG1 | J591 | MMAE | vc | Phase II | mCRPC |
| Ladiratuzumab vedotin (SGN-LIV1A) | Seattle Genetics | Hz IgG1 | LIV1 | MMAE | vc | Phase I/II | Breast cancer |
| Enapotamab Vedotin (HuMax-Axl-ADC) | Genmab | IgG1 | AXL | MMAE | vc | Phase I/II | Multiple solid tumors |
| AGS-16C3F | Astellas (Agensys) | Hu IgG2 | ENPP3 | MMAF | mc | Phase II | RCC |
| GSK-2857916 | GSK | Hz IgG1 | BCMA | MMAF | mc | Phase II | MM |
ALCL, anaplastic large cell lymphoma; HL, Hodgkin lymphoma; NHL, non-Hodgkin lymphoma; DLBCL, diffuse large B-cell lymphoma; NSCLC, non-small cell lung cancer; mCRPC, metastatic castration-resistant prostate cancer; RCC, metastatic renal cell carcinoma; MM, multiple myeloma; vc, valine–citrulline peptide linker; mc, maleimidecaproyl linker.
In this review, our recent research regarding auristatin analogues is described. The first section discusses N-terminal (P1) and C-terminal (P5) modifications and corresponding ADC data.17) The second section discusses central peptide (P2–P4) modifications, focusing on P2 and P4 SAR.18–20)
In the late 1970s, systematic research of marine life forms led to the discovery of lead structures dolastatin 1 and 2, which were extracted from Dolabella auricularia (sea-hare), a shell-less mollusk collected in Mauritius by Pettit et al.21) Both natural products exhibited promising results against the P388 leukemia cell line. Further natural product discovery efforts22,23) were engaged and in the late 1980s, the most promising compound family members, namely dolastatins 10–1524–29) were isolated and their structures elucidated.
Dolastatin 10 (6), which consists of four amino acid building blocks, dolavaline (Dov, P1), valine (Val, P2), dolaisoleuine (Dil, P3), and dolaproine (Dap, P4), and the C-terminal amine dolaphenine (Doe, P5) showed the highest cytotoxic activities against human cancer cell lines30,31) (Fig. 2). The mechanism of action responsible for these cytotoxic effects was revealed at the U.S. National Cancer Institute: dolastatin 10 binds powerfully at the tubulin vinca alkaloid binding domain, and causes cells to accumulate in metaphase arrest.32,33) Applying higher concentrations of dolastatin 10 (6) resulted in a complete disappearance of intracellular microtubules.34)
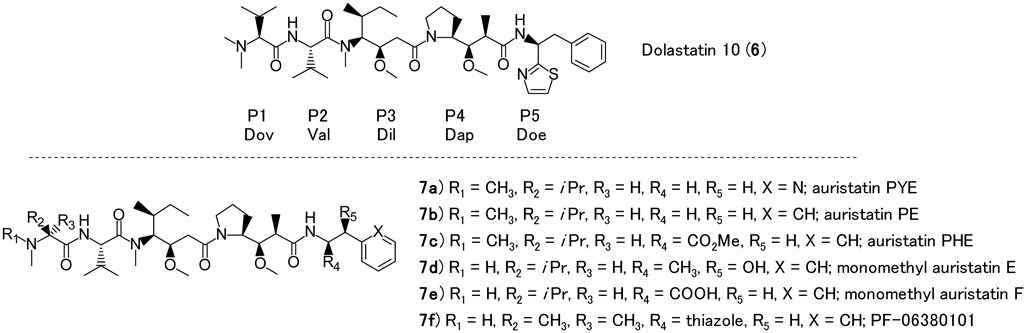
Dolastatin 10 (6) was advanced to a number of clinical studies for a variety of cancers; however, no significant clinical activity was observed at the maximum dose tolerated.35–42) The same result was obtained for water-soluble dolastatin analogs, the auristatin TZT-1027 (auristatin PE, 7b)43) (Fig. 2). As a further study to improve in vivo efficacy, new auristatin derivatives were developed, resulting in MMAE (7d) and MMAF (7e) (Fig. 2), which contain a functional handle for attachment of covalent linkers. An approach was developed to increase the therapeutic index of these compounds in clinical trials where MMAE and MMAF would be combined with a linker and cancer target-specific monoclonal antibodies as ADCs.44–49) The mechanism of action of auristatins is well suited for use in ADCs because they arrest tumor cells and at the same time are known to stimulate an immune response toward cancer cells by inducing immunogenic cell death.49) Several auristatins are used in ADCs including MMAE (7d),44–46) MMAF (7e),47) and a 2-aminoisobutyric acid auristatins derivative (PF-06380101, 7f).48) These fully synthetic dolastatin 10 derivatives should have a manufacturing advantage when compared with other ADC payloads, which are derived from natural product isolated sources.49)
Many research groups have studied auristatins with a focus mostly on the P1 and P5 subunits and these modifications have been extensively reviewed16,50) (Fig. 3). N-Terminal P1 subunit and/or C-terminal P5 subunit modifications without any alteration of the peptide core P2-P3 is a general strategy in particular where Pettit et al, Miyazaki et al., and other groups have reported their investigation on their SAR.51–53) Modification of the core central peptides (P2-P4 subunits) has not been as extensively investigated. Reported investigations into central core modifications have resulted in attenuated compound potency; however, the scope of that established SAR is not as extensive as for the P1 and P5 subunit modifications.53–56) Until recently, there has been limited reported information regarding the core central peptide modifications.

(Color figure can be accessed in the online version.)
In this section, the SAR of C-terminal peptide modification (P5) will be reviewed with a focus on hydrophilic auristatin derivatives and their activity as ADCs. The hydrophilicity of a payload in the ADC field is an important factor since more hydrophobic compounds tend to cause protein aggregates and faster drug clearance.57) Hydrophilic auristatins have been investigated and it is generally understood that increasing the hydrophilic character of an auristatin decreases the tendency to form protein aggregates58–60) and reduces the clearance rate of the corresponding ADCs from circulation.57,61,62) So as to explore the physical properties of auristatin payload charge and hydrophilicity and their impact on the potency and pharmacokinetics of subsequent ADCs, we synthesized and tested several P5 amine-containing auristatins based on the auristatin PYE (7a) scaffold, which had previously been reported to display tumor growth inhibition activity.63)
3.1.1. P5 Modification; SAR Based on in Vitro CytotoxicityOur screening paradigm for the series of synthesized compounds used in vitro cytotoxicity assays with two ADC-relevant human cancer cell lines (HER2-expressing human mammary ductal carcinoma HCC1954 cell line and HER2-negative human prostate carcinoma cell line, PC3) and an in vitro tubulin polymerization assay65,66) is shown in Table 2.
 |
aLog D values calculated using ACD/Percepta version 2015, Advanced Chemistry Development, Inc., Toronto, ON, Canada, www.acdlabs.com, 2015 (Log P: Consensus Log P, pKa: ACD/pKa Classic). bPaclitaxel stabilizes the polymerization of tubulin rather than inhibits. cNot measured. dObtained from literature.64) (Color figure can be accessed in the online version.)
The SAR of substitutions in the P5 position and correlation with log D7.4 are summarized in Fig. 4. Basically, there was a correlation between log D and cytotoxicity which can be because of cell permeability (Fig. 4C). This does not rule out inferior engagement of the tubulin target as an explanation of the lower activity of compounds having lower calculated log D7.4 values.48)

aLog D values calculated using ACD/Percepta version 2015, Advanced Chemistry Development, Inc., Toronto, ON, Canada, www.acdlabs.com, 2015 (Log P: Consensus Log P, pKa: ACD/pKa Classic). (Color figure can be accessed in the online version.)
Through the SAR in Fig. 4A, when compared to 2-pyridine-containing compound 8, the 4-pyridine-containing compound 10 loses one order of magnitude in activity and the 3-pyridine-containing compound 9 loses three orders of magnitude in activity despite being very similar in structure and hydrophilicity. We revealed that these results came from poor inhibition of tubulin polymerization 9.17) Changing the pyridine to an imidazole also resulted in some loss of potency with 1-substituted imidazole 12 being more active than either the 2-substituted 11 or the 4-substituted imidazole 13. Compounds 11 and 13 have lower calculated log D7.4 values compared with 12, which may be due to the free −NH bonds of the 2- and 4-imidazoles. This could result in lower cell permeability which can account for the observed activity. In general, compounds with aliphatic substituents 15, 16 are much less potent than those with aromatic substituents, with the exception of the morpholino derivative 14. This compound shows better-than-expected activity against HCC1954 cells despite exhibiting only moderate inhibition of tubulin polymerization.17)
The SAR of replacement of the primary or secondary amine, which are supposed to be used as linker attachment in the P5 position, is summarized in Fig. 4B. Except for compound 17, derivatives 18–20 with an aliphatic amine in the P5 position were not active because of high hydrophilicity.
As a result, some compounds, notably 2-pyridine-containing compound 8, mono methyl auristatin PYE (MMAPYE), and 17, demonstrated very potent activity with IC50 values of 1 nM in both cell lines, which are comparable to MMAE (7d) and auristatin PYE (7a) (Table 2). In this study the most cytotoxic compounds also exhibited high inhibition of tubulin polymerization. However, the reverse correlation did not hold true.16)
3.1.2. Evaluation of in Vivo Efficacy of ADC with MMAPYE (8) as PayloadThe in vitro screening experiment was followed by in vivo studies of the free auristatin molecules and the most active payload was then evaluated for use in an ADC. The MMAPYE (8) and aniline 17 were evaluated in vivo (data not shown in this paper). We identified that MMAPYE (8) was the payload with the best activity and the least toxicity in vivo16) (17 showed signs of toxicity at the 4 mg/kg dose, pronounced body weight loss). It was successfully attached to a variety of linkers then conjugated to trastuzumab for evaluation as an ADC (Fig. 5).

(Color figure can be accessed in the online version.)
The trastuzumab-linked drugs (21–23) were evaluated in vitro and in vivo with HCC1954 cells (Table 3 and Fig. 6A). Of the three ADCs tested, only the 3-(bromoacetamido) propionate-linked trastuzumab-23 showed significant efficacy with 76% tumor growth inhibition (p = 0.0103) compared versus vehicle control (Fig. 6A). It is possible that this activity is driven by the halo-acetamide-type linkage to the antibody. Compound 23 displayed antitumor effect despite the (lack of) observed in vitro activity (Table 3). MMAPYE (8) displayed activity as a free payload (Table 2) and so the in vivo data suggests that better efficacy may be obtained through optimization of the linker used to convert MMAPYE (8) into compound 23. Linking through a carbamate bond (24–27) instead of an amide functionality altered the released active metabolite. Unfortunately, compounds 24–27 showed little-to-no activity in vitro (Table 3) or in vivo (Fig. 6B) although trastuzumab-24 did appear to show some tumor growth inhibition (67%, but this was not statistically significant compared with H3-1.4-24 on day 23; p = 0.0895). Based on these results it would appear that the carbamate-containing linkers act more like traditional amide non-cleavable linkers and are not able to release the free payload by cleavage at the carbamate.
| Compound | IC50 HCC1954 (nM) | IC50 PC3 (nM) |
|---|---|---|
| Trastuzumab-21 | > 100 | > 100 |
| Trastuzumab-22 | > 100 | > 100 |
| Trastuzumab-23 | > 100 | > 100 |
| Trastuzumab-24 | > 100 | > 100 |
| Trastuzumab-25 | > 100 | > 100 |
| Trastuzumab-26 | > 100 | > 100 |
| Trastuzumab-27 | > 100 | > 100 |
| Trastuzumab-28 | 0.06 | > 100 |

(A) Female ICR/SCID mice bearing size-matched HCC1954 human breast carcinoma xenografts of 200 mm3 were given a single dose intravenously at 10 mg/kg on day 0 (n = 10). The tumor volumes for all animals on day 18 are shown with the mean and standard error. (B) Female ICR/SCID mice bearing size-matched HCC1954 human breast carcinoma xenografts of 200 mm3 were dosed intravenously at 10 mg/kg as a single dose on day 0 (n = 6). Tumor volumes for all animals are shown at day 23 with the mean and standard error.
On the other hand, cleavable ADCs, such as a cleavable mc-Val-Cit-Paba linker,45,67) resulted in significant tumor regression. In the HCC1954 human breast carcinoma orthotopic xenograft model, trastuzumab-28 showed similar efficacy to Kadcyla® (an approved ADC for Her2 positive breast cancer)68) (Fig. 7). Both trastuzumab-28 and Kadcyla® showed potent antitumor effect (p < 0.0001) compared versus control ADC (Fig. 7A). Treatment with a single dose of 5 mg/kg of trastuzumab-28 or Kadcyla® also resulted in statistically significant tumor regression (77.7% and 63.0%, respectively; p < 0.0001) (Fig. 7B). This result is very encouraging; we were able to show significant tumor growth inhibition with a cleavable linker when using MMAPYE (8) as part of an ADC.

Human breast carcinoma HCC1954 cells (3 × 106 cells per mouse) were implanted into the mammary fatpad of female CB17/SCID mice. Treatment with trastuzumab-36 and Kadcyla® at 5 mg/kg as a single dose was started when tumors reached 200 mm3 (n = 10). cHmLys.1c3.G2k-36 is the nonbinding control ADC and the vehicle is 20 mM histidine/5% trehalose, pH 5.2. (A) Graph shows mean tumor volume over time with standard error for each cohort. (B) Individual tumor volumes on day 21 in each cohort with mean and standard error. (Color figure can be accessed in the online version.)
To evaluate the effect of payload 8 on aggregation, ADCs containing a cleavable or noncleavable drug-linker were generated with increasing DAR. This was done to evaluate whether increased drug hydrophilicity would cause less antibody aggregation. The study builds on previous reports that aggregation of auristatins ADCs tends to increase with increasing DAR.61,62,69–71) Since in our experience human immunoglobulin G (IgG)1 antibodies such as trastuzumab are not typically as prone to aggregation as human IgG2 antibodies, the drug-linkers were conjugated to two human IgG2 monoclonal antibodies (hIgG2-A and hIgG2-B) via interchain cysteine residues for evaluation with DARs ranging from 2 to 8. The level of antibody aggregation was measured by size exclusion chromatography (SEC). Conjugation to mc-Val-Cit-Paba-8 (compound 28) caused a modest increase in soluble aggregate in the case of hIgG2-A (Fig. 8A) or a slight decrease in measured soluble aggregate in the case of hIgG2-B (Fig. 8B), which can be explained by partial precipitation of aggregate present prior to conjugation. The changes in aggregation with increasing DAR were less pronounced than in previous reports for antibodies conjugated to mc-Val-Cit-Paba-MMAE.66,68) Conjugation to mc-8 (compound 22) resulted in no increase in soluble aggregate as the DAR increased. Conjugation of the same druglinkers to trastuzumab (DAR 2 − 8) did not result in more than 5% soluble aggregate for either drug-linker.17) These results support our hypothesis that the propensity of an ADC to develop soluble aggregate can be mitigated by increasing the hydrophilicity of the payload portion of the drug-linker. To confirm this contention, we compared the retention time of a trastuzumab-28 conjugate by hydrophobic interaction chromatography (HIC) with that reported in published data for a trastuzumab–vcMMAE conjugate72) using the same experimental conditions and analytical method.17) The results show that trastuzumab-28 is retained less than trastuzumab-vcMMAE under the same assay conditions (Fig. 9), indicating that the payload is more hydrophilic than MMAE.

Aggregation was measured by SEC.

Overall, the results of this study show that using MMAPYE (8) as part of an ADC may enable us to overcome some of the problems seen when using more hydrophobic molecules. This is encouraging and opens up the possibility to study other auristatins containing heterocycles, including methyl imidazoles, thiazoles, thiophenes, oxazoles, and indoles as potential ADC payloads.
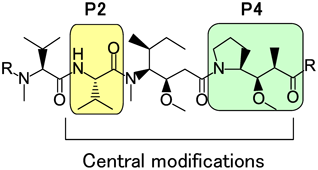
3.2. Central Peptide ModificationIn this section, novel modifications to the central peptide section of the auristatin backbone are described (Fig. 10). The SAR described in the chemical literature is significantly more limited16,50,53–56) for the P2–P4 modifications compared with the terminal amino acid variants. For the P2 subunit, limited substitution of the natural valine residue with leucine or isoleucine was previously reported without a significant impact in compound potency.53) Regarding the P4 subunit, analogues with mannose- and glucose-derived sugar amino acids as replacements for the Dap portion were examined by Gajula et al.54,55) whereas hydroxyl-, methoxy-, and amino-substituents on the pyrrolidine ring were described by Park et al.56)
(Color figure can be accessed in the online version.)
We decided to investigate modifications of the central core in greater depth than had been reported at that time to gain a better understanding of the potency tolerance of these modifications and to explore possibilities of creating linkage sites to auristatin analogs through the central units. Our goal was to create novel compounds with enhanced properties for use in ADCs.
First, we created a set of auristatin compounds containing heteroatoms in the central core amino acids and evaluated them to determine whether heteroatom introduction could be an effective approach to control cytotoxic activity and serve as a handle for new linker attachment at P2 or P4 instead of at P1 or P5.18,19)
3.2.1. P2 ModificationWe investigated SAR for analogues containing heteroatoms and other non-natural amino acids in the P2 position.18) The functional groups chosen for incorporation in the P2 amino acid unit were amine, azide, hydroxyl, and thiol. From a membrane permeability standpoint, P2 modified analogues with L-phenylalanine (Phe) methyl ester at the P5 unit were compared (Fig. 11).

(Color figure can be accessed in the online version.)
Through their resulting cytotoxic activities in in vitro tumor cell proliferation assays using HCC1954 and PC3 cell lines, compounds 29, 30, and 31 emerged as lead compounds with in vitro activities on the order of 10−11–10−10 M 50% growth inhibition (GI50) values (Fig. 11, Table 4). Notably, compound 31 contained a free branched amine in the P2 side chain that at physiological pH would have a positive charge. Despite this charge, compound 31 was active compared with other amine derivatives containing unbranched free amino units in the P2 side chain position. An alkyne functionality in the P2 side chain displayed similar activity in vitro to the azide-containing compounds. An alcohol unit in the P2 position lost one order of magnitude of cytotoxic activity compared with the azide-containing compounds. A cysteine unit in the P2 position was not as active compared with other P2-substituted compounds possibly due to the formation of disulfide dimers that can occur spontaneously under conditions of purification and the cytotoxicity assay (data not shown). A carboxylic acid in the P2 position was not active against any of the cell lines tested.
 | |||||
|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | GI50 HCC1954 (nM)a | GI50 PC3 (nM)a |
| 29 | (S)-CH(N3)CH3 | CO2Me | CH2Ph | 0.041 | 0.093 |
| 30 | CH2N3 | CO2Me | CH2Ph | 0.036 | 0.11 |
| 31 | (S)-CH(NH2)CH3 | CO2Me | CH2Ph | 0.076 | 0.54 |
| 32 | (S)-CH(N3)CH3 | CO2H | CH2Ph | >1000 | >1000 |
| 33 | (S)-CH(NH2)CH3 | CO2H | CH2Ph | 2.5 | 11 |
| 34 | (S)-CH(N3)CH3 | H | CH2Ph | 3.1 | 4.7 |
| 35 | (S)-CH(N3)CH3 | Me | (S)-CH(OH)Ph | 34 | 50 |
| 36 | (S)-CH(N3)CH3 | H | CH2-Ph(4-Cl) | 33 | 40 |
| 37 | (S)-CH(N3)CH3 | H | CH2-Ph(2-Cl) | 13 | 34 |
| 38 | (S)-CH(N3)CH3 | H | CH2-2Py | 3.7 | 5.1 |
| 39 | (S)-CH(N3)CH3 | CONH2 | CH2Ph | 0.4 | 1.7 |
| 40 | (S)-CH(N3)CH3 | CONHtBu | CH2Ph | 0.4 | 1.2 |
| 41 | (S)-CH(N3)CH3 | CO2tBu | CH2Ph | 0.089 | 0.045 |
| 42 | (S)-CH(N3)CH3 | Tetrazole | CH2Ph | 27 | 41 |
| 43 | (S)-CH(N3)CH3 | 2-Thiazole | CH2Ph | 18 | 33 |
a Results are average of 2 independent triplicate runs with compound purity >90%. (Color figure can be accessed in the online version.)
The SAR with the combination of P2 and P5 subunit is described17) (Table 4).
Compounds 32, 33 were synthesized with a phenylalanine in the P5 position. As expected, these were less active compared versus their corresponding methyl ester counterparts probably due to reduced membrane permeability. Despite the poor membrane permeability, these compounds may still have usefulness as targeted ADC payload because antibodies have the ability to release the payload inside the cells, such as MMAF (7e)-derived ADCs.
P2-azide modified auristatins with phenylethylamine (34), norephedrine (35), 4-chlorophenylethylamine (36), and 2-chlorophenylethylamine (37) present in the P5 position resulted in a loss of potency compared with the phenylalanine-based P5 derivatives. These results did not follow the previous known results where phenylethylamine derivatives are usually equipotent with phenylalanine and dolaphenine. In our previous SAR with P5, we showed that MMAPYE (8, Table 2) had high in vitro activity and lower aggregation as an ADC. Based on this, the PYE P5 was introduced to P2 azide compounds. The compound 38 lost potency compared with MMAPYE (8) (<1 nM), but still had single-digit nanomolar GI50 values.
Since the phenylalanine P5 was required for activity of the P2-azide compounds, we explored different phenylalanine derivatives including Phe-carboxamide (39), Phe-tert-butyl amide (40), Phe-tert-butyl ester (41), and two carbonyl type mimics, a tetrazole (42) and a thiazole (43). Compounds 39–41, which have an ester or amide at P5, showed potent activity in vitro. The presence of bigger, bulkier P5 groups Phe-tert-butyl amide (40) and Phe-tert-butyl ester (41) gave similar results to the smaller amide and ester Phe-carboxamide (39) and Phe-methyl ester (29). On the other hand, the carbonyl type mimics 42 and 43 showed loss of potency. These compounds do inhibit tubulin polymerization in a biochemical assay,18) suggesting that other factors for in vitro potency are at play other than simply tubulin polymerization inhibition.
Thus through SAR study of P2 modifications, we revealed that the most active compounds contained azides in the P2 unit with a phenylalanine-derived P5 subunit.
3.2.2. P4 ModificationAfter investigating SAR on P2 subunit, our interest moved on P4-modified analogues containing linkage handles or heteroatoms. Auristatin analogues with an azide-substituted central P4 amino acid in combination with azide-containing P2 substitutions were synthesized.
The in vitro potency of each compound with respective P1, P2, and P4 unit modifications were summarized19) (Fig. 12). Each compound was evaluated in an in vitro cell proliferation assay using an acute myeloid leukemia cell line (MOLM13). All compounds demonstrated similar GI50 values (within one log). Based on our previous studies evaluating P2 modifications,18) we held the aromatic P5 ester units unchanged while examining the SAR of P4 azide introduction. Compound 44, containing an azide only in the P4 central peptide, demonstrated similar activity in vitro compared with MMAE (7d). Next, we explored combinations of both P4 and P2 azide modifications. Compound 45, with P2-azide and a MeVal unit in the P1 position, showed a similar GI50 value compared with 44. Compound 46, with P2 and P4 azide modifications and Dov in the P1 position, showed great cytotoxic activity (GI50 = 0.057 nM) compared with compound 45. The potency in themselves could be due to many factors such as binding to the tubulin dimer, conformation of the pentapeptide, or changes in active transport.48,49) Based on a critical hydrogen bond network between pentapeptide and amino acid residue in tubulin and the cis Val-Dil amide bond and trans Dil-Dap amide bond geometries, which were revealed in PF-06380101 (7f) tubulin cocrystal structure data,48) it might be difficult for azide groups to interact with amino acid residues of tubulin to boost activity. Rather, the azide group might tend to force the pentapeptide conformation and give favorable conformation to yield these suitable interactions with amino acid residues of tubulin.

(Color figure can be accessed in the online version.)
As well as discovering new auristatin analogues containing azides, we also confirmed that azide groups introduced into the P2 and P4 subunits demonstrate some possibility for serving as handles for linker attachment. Compounds with non-cleavable linkers 49, 50 were prepared using click chemistry73) (Fig. 13). These payloads incorporating azide groups could serve as linker attachment sites for other non-cleavable or cleavable linkers. Thus these compounds with azide in P2 and/or P4 are promising payloads for preparation of next generation type of novel drug linker at the P2 or P4 position.

(Color figure can be accessed in the online version.)
We provided new directions toward next generation auristatin derivatives. We revealed that introduction of hydrophilic character in the P5 subunit can control auristatin properties and generate an efficacious ADC17) and the introduction of heteroatoms, especially azide in central core, can modulate or enhance cytotoxic activity while introducing a P2 or P4 linker attachment site (instead of the more common P1 or P5 linkage approaches) in the preparation of ADCs.18,19)
Through study of P5 modified analogues, the in vitro cytotoxicity screen showed that some weakly basic aromatic side-chain groups were tolerated with minimal loss of activity. Pyridines and anilines retained activity but the more strongly basic and aliphatic groups such as 2- and 4-imidazoles had diminished cytotoxic activity. Therefore, compound 8 was identified as best hydrophilic auristatin based on in vitro and in vivo studies.
ADCs with an enzyme-cleavable mc-Val-Cit-Paba linker, such as trastuzumab-28, showed tumor regression when tested in vivo compared with ADCs using noncleavable linkers. This result demonstrates that payloads containing basic nitrogen groups can be successfully used as part of an ADC when combined with a cleavable linker.
On the other hand, ADCs with noncleavable drug-linkers showed no activity in vitro and only moderate or no efficacy in vivo. Carbamate linkers showed similar behavior as noncleavable ADCs, which appears not to release the free drug after internalization under the conditions tested. Further studies assessing the metabolites generated by these linkers are necessary to confirm our hypothesis where these ADCs would release free drug after intracellular degradation of the carbamate linkage. For instance, it may be possible to increase the release of free drug by introducing a spacer to move the carbamate away from the N-methyl group on the auristatin P1, making it more accessible for enzyme degradation.
A study of the relation between aggregate and DAR was carried out on a human IgG2 antibody since, in our experience, they often have a greater propensity to aggregate than do human IgG1 antibodies. The degree of protein aggregation did not increase during conjugation of mc-8 (22) or mc-Val-Cit-Paba-8 (28) in an increasing-DAR titration study. This payload may be a good candidate to use with an antibody that has a tendency to aggregate and may help control the properties of the resulting ADC.
In terms of central peptide subunit, through studies of SAR, some new auristatin analogues with azide groups at P2 and/or P4 were obtained with great potency. This great potency in itself might come from the favorable conformation caused by azide groups to allow suitable interactions with amino acid residues of tubulin. In addition, with non-cleavable linker in ADC, P2 and P4 subunit might be suitable positions as linker attachment sites because after lysosomal catabolism to release the payload species,47) catabolites substituted at P2 or P4 position; these P2 and P4 substitutions would not interrupt the critical hydrogen bond network but would rather control suitable conformation. Using P2 or P4 linker in ADC might be a useful method to optimize ADC. Additional research is needed in this chemistry through co-crystal structure study.
Toward creation of next generation ADCs, further study is ongoing and we will investigate ADCs with our payloads and linkers.
We gratefully acknowledge the work of co-workers of the Protein Technology group and appreciate all Agensys ADC team members.
The authors declare no conflict of interest.