Experimental
UV-vis spectra were recorded on a JASCO Ubest-55 or JASCO V-650 instrument. IR spectra were measured on a PerkinElmer, Inc. FT-IR spectrometer, model Paragon 1000 or Horiba FT-IR spectrometer, model FT-720. NMR spectra were obtained on a Varian Mercury-300, a VXR-500 or a Brucker Avance III 600 superconducting FT-NMR spectrometer. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane as an internal reference (CDCl3: δ = 0 ppm for 1H) or residual solvent signals (CDCl3: δ = 77 ppm for 13C). Mass spectra were taken on a Thermo Fisher Scientific Exactive spectrometer. Column chromatography was performed using Kanto Silica Gel 60 N (spherical, neutral). All reagents were used as obtained commercially unless otherwise noted.
General Procedure for the Synthesis of C2-Elongeted Conjugate Ester by Horner–Wadsworth–Emmons Reaction (GP1)To a stirred solution of triethyl phosphonoacetate (C2-phosphonate, 2.5–5.0 eq.) in dry tetrahydrofuran (THF) (0.2 M/phosphonate) was added a solution of NaN(TMS)2 (2.5–5.0 eq.) or n-BuLi (2.5–5.0 eq.) at 0°C, and the mixture was stirred at the same temperature for 15 min. To this mixture a solution of aldehyde (1 eq.) in THF (1 M/aldehyde) was added, and the reaction mixture was stirred at room temperature (r.t.) until the reaction was completed. The reaction was quenched with a saturated aqueous solution of NH4Cl, and then extracted with ethyl acetate. The combined extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel to afford the one double bond elongated conjugate ester. In the case of A2 analogs, sodium hydride (NaH) (1.5 eq., 60% suspension in mineral oil) was used as a base instead of NaN(TMS)2.
General Procedure for the Synthesis of C5-Elongeted Conjugate Ester by Horner–Wadsworth–Emmons Reaction (GP2)To a stirred solution of triethyl 3-methyl-4-phosphonocrotonate (C5-phosphonate, 3.0 eq.) and DMPU (4 eq.) in dry THF (0.2 M/aldehyde), n-BuLi (3.0 eq.) was added at 0°C. The resulting mixture was stirred at the same temperature for 20 min and then cooled to −78°C. To this mixture a solution of aldehyde (1 eq.) in THF (1 M/aldehyde) was slowly added, and the reaction mixture was allowed to come to r.t. and then stirred for overnight. The reaction was quenched with a saturated aqueous solution of NH4Cl, and then extracted with ethyl acetate. The combined extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel to afford the two double bonds elongated conjugate ester as a stereoisomeric mixture (E : Z = 9 : 1 to 18 : 1). The mixture was used for the next reaction without further separation of stereoisomers.
General Procedure for the Conversion from Ester to Aldehyde (GP3)In the case of A1-analogs, to a stirred suspension of LiAlH4 (2.0 eq.) in dry Et2O (0.2 M/aldehyde) was added a solution of ester (1 eq.) in Et2O (0.2 M/ester) at 0°C, and the resulting mixture was stirred at the same temperature for 1 h. To this mixture aqueous silica gel (H2O/SiO2 = 1 : 5) was added and the resulting mixture was stirred at r.t. for 1 h. The reaction mixture was filtered through Celite with Et2O, dried over Na2SO4, and concentrated in vacuo. The resultant allyl alcohol was used for the next MnO2 oxidation step without further purification.
In the case of A2-analogs, to a solution of ester (1 eq.) in THF (0.1 M/ester) was added DIBAL (3.5 eq., 1.0 M in hexane) dropwise at −78°C, and the mixture was stirred at the same temperature for 2 h. The mixture was quenched with H2O/SiO2, and was stirred at r.t. for 30 min. The resulting mixture was filtered through Celite with Et2O, and evaporated in vacuo. The resultant allyl alcohol was used for the next MnO2 oxidation step without further purification.
To a solution of ally alcohol in Et2O (0.04 M) was added MnO2 (10 times weight relative to ester) at r.t. and was stirred for 14–18 h. The resulting mixture was filtered through Celite with Et2O, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel to afford the aldehyde as pure E stereoisomer.
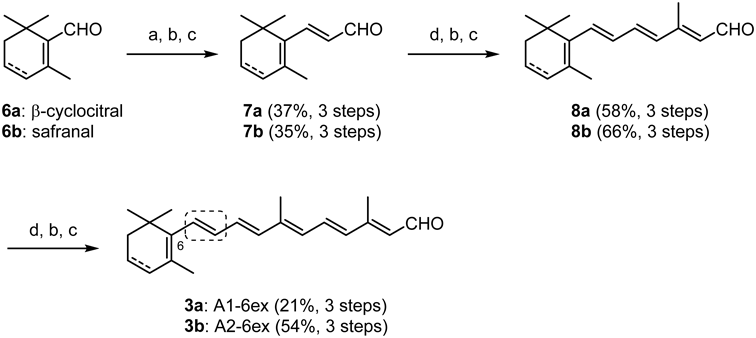
(E)-3-(2,6,6-Trimethylcyclohexen-1-yl)-2-propen-1-al (7a)According to GP1, ethyl (E)-3-(2,6,6-trimethylcyclohexen-1-yl)-2-propenate (1.37 g, 94%) was obtained from β-cyclocitral (6a: 1.00 g, 6.57 mmol), triethyl phosphonoacetate (7.36 g, 32.9 mmol) and n-BuLi (20.5 mL, 1.57 M in hexane, 32.2 mmol). Eluent: hexane/EtOAc = 9 : 1. According to GP3, 7a (428 mg, 37%, in 3 steps from 6a) was obtained from ethyl (E)-3-(2,6,6-trimethylcyclohexen-1-yl)-2-propenoate (1.37 g, 6.18 mmol), LiAlH4 (469 mg, 12.4 mmol) and MnO2 (85% purity, 14.0 g, 137 mmol). Eluent: hexane/Et2O = 4 : 1. Spectral data were identical with those of the literature.26)
7a: Pale yellow oil. IR (CHCl3) νmax cm−1: 2928, 1676, 1605. 1H-NMR (300 MHz, CDCl3) δ: 9.51 (1H, d, J = 7.8 Hz), 7.26 (1H, d, J = 16.2 Hz), 6.17 (1H, dd, J = 16.2, 7.8 Hz), 2.10 (2H, t, J = 6.0 Hz), 1.79 (3H, s), 1.70–1.53 (2H, m), 1.53–1.47 (2H, m), 1.09 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 194.95, 152.99, 139.80, 136.25, 132.79, 40.17, 34.26, 34.24, 28.90, 22.02, 18.94.
(E)-3-(2,6,6-Trimethylcyclohexa-1,3-dien-1-yl)-2-propen-1-al (7b)According to GP1, ethyl (E)-3-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2-propenate (11.9 g, 87%) was obtained from safranal (6b: 9.33 mL, 60.0 mmol), triethyl phosphonoacetate (20.2 g, 90.0 mmol) and NaH (60%, 3.60 g, 90.0 mmol). Eluent: hexane/Et2O = 20 : 1. According to GP3, 7b (3.74 g, 35%, in 3 steps from 6b) was obtained from ethyl (E)-3-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2-propenate (11.9 g, 53.8 mmol), LiAlH4 (5.11 g, 135 mmol) and MnO2 (85% purity, 119 g, 1.16 mol). Eluent: hexane/Et2O = 10 : 1
7b: Yellow solid. IR (CHCl3) νmax cm−1: 2962, 1670, 1607. 1H-NMR (300 MHz, CDCl3) δ: 9.54 (1H, d, J = 7.8 Hz), 7.26 (1H, d, J = 16.2 Hz), 6.26 (1H, dd, J = 16.2, 7.8 Hz), 5.99–5.89 (2H, m), 2.15 (2H, d, J = 3.3 Hz), 1.94 (3H, s), 1.11 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 194.35, 151.23, 135.69, 135.09, 131.62, 129.68, 129.64, 40.16, 34.01, 26.53, 20.41. High resolution electrospray ionization (HR-ESI) MS Calcd for C12H17O [M + H]+: 177.1274. Found: 177.1273.
(2E,4E,6E)-3-Methyl-7-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6-heptatrien-1-al (8a)According to GP2, ethyl (E)-3-methyl-7-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6-heptatrienoate (628 mg, 90%) was obtained from 7a (428 mg, 2.40 mmol), triethyl 3-methyl-4-phosphonocrotonate (1.77 g, 7.20 mmol), n-BuLi (4.50 mL, 1.57 M in hexane, 7.07 mmol) and DMPU (1.10 mL, 9.59 mmol). Eluent: hexane/EtOAc = 9 : 1. According to GP3, 8a (360 mg, 58%, in 3 steps from 7a) was obtained from ethyl (2E,4E,6E)-3-methyl-7-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6-heptatrienoate (628 mg, 2.18 mmol), LiAlH4 (165 mg, 4.35 mmol) and MnO2 (85% purity, 6.30 g, 61.6 mmol). Eluent: hexane/Et2O = 4 : 1. This compound is known and main signals of 1H-NMR were reported in the literature.27)
8a: Pale yellow oil. IR (CHCl3) νmax cm−1: 2932, 1656, 1594. 1H-NMR (300 MHz, CDCl3) δ: 10.10 (1H, d, J = 8.1 Hz), 6.82 (1H, dd, J = 15.6, 10.5 Hz), 6.46 (1H, d, J = 15.6 Hz), 6.31 (1H, d, J = 15.6 Hz), 6.26 (1H, dd, J = 15.6, 10.5 Hz), 5.95 (1H, d, J = 8.1 Hz), 2.31 (3H, s), 2.05 (2H, t, J = 6.3 Hz), 1.75 (3H, s), 1.64–1.56 (2H, m), 1.52–1.44 (2H, m), 1.05 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 191.16, 154.91, 137.53, 137.25, 133.17, 132.56, 132.40, 128.95, 39.71, 34.15, 33.39, 28.90, 21.79, 19.05, 13.00.
(2E,4E,6E)-3-Methyl-7-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6-heptatrien-1-al (8b)According to GP2, ethyl (2E,4E,6E)-3-methyl-7-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6-heptatrienoate (7.98 g, 95%) was obtained from 7b (5.20 g, 29.5 mmol), triethyl 3-methyl-4- phosphonocrotonate (11.6 g, 44.2 mmol), n-BuLi (28.0 mL, 1.58 M in hexane, 44.2 mmol) and DMPU (7.11 mL, 59.0 mmol). Eluent: hexane/EtOAc = 30 : 1.
According to GP3, 8b (4.72 g, 66%, in 3 steps from 7b) was obtained from ethyl (2E,4E,6E)-3-methyl-7-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6-heptatrienoate (7.98 g, 27.9 mmol), DIBAL (100 mL, 1.0 M in hexane, 100 mmol) and MnO2 (85% purity, 79.8 g, 780 mmol). Eluent: hexane/Et2O = 4 : 1.
8b: Yellow solid. IR (CHCl3) νmax cm−1: 3013, 1651, 1592. 1H-NMR (300 MHz, CDCl3) δ: 10.11 (1H, d, J = 8.1 Hz), 6.83 (1H, dd, J = 15.6, 9.9 Hz), 6.48 (1H, d, J = 15.6 Hz), 6.36 (1H, dd, J = 15.6, 9.9 Hz), 6.35 (1H, d, J = 15.6 Hz), 5.96 (1H, d, J = 8.1 Hz), 5.87 (1H, d, J = 9.6 Hz), 5.79 (1H, dt, J = 9.6, 4.2 Hz), 2.31 (s, 3H), 2.10 (2H, dd, J = 4.2, 0.9 Hz), 1.90 (3H, s), 1.06 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 191.00, 154.66, 137.48, 137.42, 135.86, 133.45, 131.81, 129.84, 129.51, 128.96, 126.43, 39.92, 33.86, 26.71, 20.39, 12.92. HR-ESIMS Calcd for C17H23O [M + H]+: 243.1743. Found: 243.1745.
(2E,4E,6E,8E,10E)-3,7-Dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaen-1-al (A1-6ex, 3a)According to GP2, ethyl (2E,4E,6E,8E,10E)-3,7-dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaenoate (459 mg, 93%) was obtained from 8a (360 mg, 1.39 mmol), triethyl 3-methyl-4-phosphonocrotonate (1.02 g, 4.16 mmol), n-BuLi (2.60 mL, 1.57 M in hexane, 4.08 mmol) and DMPU (0.630 mL, 5.23 mmol). Eluent: hexane/EtOAc = 9 : 1. According to GP3, 3a (99.0 mg, 21%, in 3 steps from 8a) was obtained from ethyl (2E,4E,6E,8E,10E)-3,7- dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaenoate (358 mg, 1.01 mmol), LiAlH4 (77.0 mg, 2.02 mmol) and MnO2 (85% purity, 3.61 g, 35.3 mmol). Eluent: hexane/Et2O = 4 : 1.
3a: Orange solid. UV-vis (EtOH) λmax 403 nm (ε = 54300). nm. IR (CHCl3) νmax cm−1: 2931, 1651, 1602, 1561. 1H-NMR (300 MHz, CDCl3) δ: 10.10 (1H, d, J = 8.4 Hz), 7.12 (1H,dd, J = 15.0, 11.4 Hz), 6.50 (1H, dd, J = 15.0, 9.9 Hz), 6.37 (1H, d, J = 15.0 Hz), 6.29 (2H, d, J = 15.0 Hz), 6.22 (1H, d, J = 11.4 Hz), 6.19 (1H, dd, J = 15.3, 9.9 Hz), 5.98 (1H, d, J = 8.1 Hz), 2.33 (3H, s), 2.03 (2H, t, J = 6.0 Hz), 2.02 (3H, s), 1.74 (3H, s), 1.64–1.58 (2H, m), 148–1.45 (2H, m), 1.04 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 191.06, 154.70, 141.15, 137.47, 134.86, 134.71, 133.52, 133.38, 132.44, 132.19, 131.04, 130.24, 129.07, 39.72, 34.17, 33.30, 28.92, 21.79, 19.15, 13.08. HR-ESIMS Calcd for C22H31O [M + H]+: 311.2369. Found: 311.2368.
(2E,4E,6E,8E,10E)-3,7-Dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaen-1-al (A2-6ex, 3b)According to GP2, ethyl (2E,4E,6E,8E,10E)-3,7-dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaenoate (2.99 g, quant.) was obtained from 8b (1.94 g, 8.00 mmol), triethyl 3-methyl-4-phosphonocrotonate (3.75 g, 12.0 mmol), n-BuLi (7.59 mL, 1.58 M in hexane, 12.0 mmol) and DMPU (1.93 mL, 16.0 mmol). Eluent: hexane/EtOAc = 30 : 1. According to GP3, 3b (1.32 g, 54%, in 3 steps from 8b) was obtained from ethyl (2E,4E,6E,8E,10E)-3,7-dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaenoate (2.99 g, 8.49 mmol), DIBAL (29.7 mL, 1.0 M in hexane, 29.7 mmol) and MnO2 (85% purity, 29.9 g, 292 mmol). Eluent: hexane/Et2O = 5 : 1. Spectral data were shown in the literature.20)
(2E,4E,6E)-5-Methyl-7-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6-heptatrien-1-al (10a)According to GP1, ethyl (2E,4E,6E)-5-methyl-7-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6- heptatrienoate (1.20 g, 99%) was obtaind from β-ionylideneacetaldehyde23) (9a: 916 mg, 4.20 mmol), NaN(TMS)2 (10.5 mL, 1.0 M in THF, 10.5 mmol) and triethyl phosphonoacetate (2.40 g, 10.5 mmol). Eluent: hexane/Et2O = 8 : 1. According to GP3, 10a (586 mg, 62%, in 3 steps from 9a) was obtained from ethyl (2E,4E,6E)-5-methyl-7-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6-heptatrienoate (1.09 g, 3.78 mmol), LiAlH4 (287 mg, 7.57 mmol) and MnO2 (85% purity, 11.0 g, 107 mmol). Eluent: hexane/Et2O = 4 : 1. Spectra data were identical with those of the literature.26)
10a: Pale yellow oil. IR (CHCl3) νmax cm−1: 2932, 1663, 1590. 1H-NMR (300 MHz, CDCl3) δ: 9.61 (1H, d, J = 8.1 Hz), 7.53 (1H, dd, J = 15.0, 12.0 Hz), 6.50 (1H, d, J = 15.9 Hz), 6.29 (1H, d, J = 12.0 Hz), 6.20 (1H, d, J = 15.9 Hz), 6.15 (1H, dd, J = 15.0, 8.1 Hz), 2.09 (3H, s), 2.04 (2H, t, J = 6.0 Hz), 1.72 (3H, s), 1.69–1.58 (2H, m), 1.51–1.42 (2H, m), 1.04 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 193.95, 148.29, 147.01, 137.60, 136.64, 132.73, 131.82, 130.95, 128.65, 39.80, 34.48, 33.43, 29.18, 22.01, 19.34, 13.37.
(2E,4E,6E)-5-Methyl-7-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6-heptatrien-1-al (10b)According to GP1, ethyl (2E,4E,6E)-5-methyl-7-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6-heptatrienoate (1.74 g, 79%) was obtained from 9b24) (1.66 g, 7.69 mmol), triethyl phosphonoacetate (2.59 g, 11.5 mmol) and NaH (60%, 0.46 g, 11.5 mmol). Eluent: hexane/Et2O = 15 : 1. According to GP3, 10b (0.914 g, 49%, in 3 steps from 9b) was obtained from ethyl (2E,4E,6E)-5-methyl-7-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6-heptatrienoate (1.74 g, 6.07 mmol), DIBAL (21.2 mL, 1.0 M in hexane, 21.2 mmol) and MnO2 (85% purity, 17.4 g, 170 mmol). Eluent: hexane/Et2O = 8 : 1.
10b: Yellow solid. IR (CHCl3) νmax cm−1: 3018, 1667, 1609. 1H-NMR (300 MHz, CDCl3) δ: 9.62 (1H, d, J = 8.1 Hz), 7.55 (1H, dd, J = 15.0, 11.7 Hz), 6.51 (1H, d, J = 16.2 Hz), 6.34 (1H, d, J = 16.2 Hz), 6.33 (1H, d, J = 11.7 Hz), 6.18 (1H, dd, J = 15.0, 8.1 Hz), 5.87 (1H, d, J = 9.6 Hz), 5.79 (1H, dt, J = 9.6, 4.5 Hz), 2.12–2.08 (5H, m), 1.89 (3H, s), 1.05 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 193.60, 147.83, 146.53, 137.78, 135.67, 131.18, 130.76, 129.76, 128.79, 127.80, 126.20, 39.86, 33.98, 26.71, 20.32, 12.99. HR-ESIMS Calcd for C17H23O [M + H]+: 243.1743. Found: 243.1741.
(2E,4E,6E,8E,10E)-3,9-Dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaen-1-al (A1-10ex, 4a)According to GP2, ethyl (2E,4E,6E,8E,10E)-3,9-dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaenoate (820 mg, 96%) was obtaind from 10a (586 mg, 2.40 mmol), triethyl 3-methyl-4-phosphonocrotonate (1.77 g, 7.19 mmol), n-BuLi (4.50 mL, 1.57 M in hexane, 7.07 mmol) and DMPU (1.10 mL, 9.59 mmol). Eluent: hexane/Et2O = 8 : 1. According to GP3, 4a (235 mg, 36%, in 3 steps from 10a) was obtained from ethyl (2E,4E,6E,8E,10E)-3,9-dimethyl-11-(2,6,6-trimethyl-cyclohexen-1-yl)-2,4,6,8,10-undecapentaenoate (709 mg, 2.00 mmol), LiAlH4 (152 mg, 4.01 mmol) and MnO2 (85% purity, 7.10 g, 69.4 mmol). Eluent: hexane/Et2O = 4 : 1.
4a: Orange solid. UV-vis (EtOH) λmax 403 nm (ε = 54000). IR (CHCl3) νmax cm−1: 2926, 1651, 1599,1561. 1H-NMR (300 MHz, CDCl3) δ: 10.10 (1H, d, J = 8.1 Hz), 6.99–6.78 (2H, m), 6.35 (1H, d, J = 15.0 Hz), 6.34 (1H, dd, J = 15.0, 10.5 Hz), 6.27 (1H, d, J = 15.6 Hz), 6.15 (1H, d, J = 10.5 Hz), 6.14 (1H, d, J = 15.6 Hz), 5.96 (1H, d, J = 8.1 Hz), 2.30 (3H, s), 2.02 (2H, t, J = 6.3 Hz), 1.99 (3H, s), 1.72 (3H, s), 1.63–1.54 (2H, m), 1.50–1.42 (2H, m), 1.03 (6H, s). 13C-NMR (75 MHz, CDCl3) δ: 191.09, 154.56, 139.51, 137.70, 137.23, 136.92, 134.46, 134.23, 131.59, 130.18, 129.68, 129.14, 128.81, 39.59, 34.26, 33.13, 28.96, 21.76, 19.19, 12.98, 12.79. HR-ESIMS Calcd for C22H31O [M + H]+:311.2369. Found: 311.2369.
(2E,4E,6E,8E,10E)-3,9-Dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaen-1-al (A2-10ex, 4b)According to GP2, ethyl (2E,4E,6E,8E,10E)-3,9-dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaenoate (714 mg, quant.) was obtained from 10b (450 mg, 1.86 mmol), triethyl 3-methyl-4-phosphonocrotonate (746 mg, 2.79 mmol), n-BuLi (1.74 mL, 1.58 M in hexane, 2.79 mmol) and DMPU (0.448 mL, 3.71 mmol). Eluent: hexane/EtOAc = 30 : 1. According to GP3, 4b (127 mg, 22%, in 3 steps from 10b) was obtained from ethyl (2E,4E,6E,8E,10E)-3,9-dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaenoate (714 mg, 2.02 mmol), DIBAL (7.08 mL, 1.0 M in hexane, 7.08 mmol) and MnO2 (85% purity, 7.14 g, 69.8 mmol). Eluent: hexane/Et2O = 5 : 1. Spectral data were shown in the literature.20)
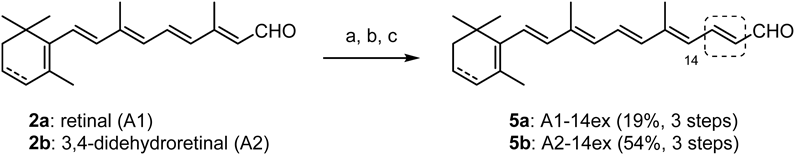
(2E,4E,6E,8E,10E)-5,9-Dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaen-1-al (A1-14ex, 5a)According to GP1, ethyl (2E,4E,6E,8E,10E)-5,9-dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaenoate (565 mg, 96%) was obtaind from retinal25) (2a: 477 mg, 1.59 mmol), triethyl phosphonoacetate (1.07 g, 4.47 mmol) and NaN(TMS)2 (4.47 mL, 1.0 M in THF, 4.47 mmol). Eluent: hexane/EtOAc = 9 : 1.
According to GP3, 5a (82.0 mg, 19%, in 3 steps from 2a) was obtained from ethyl (2E,4E,6E,8E,10E)-5,9-dimethyl-11-(2,6,6-trimethylcyclohexen-1-yl)-2,4,6,8,10-undecapentaenoate (466 mg, 1.26 mmol), LiAlH4 (96.0 mg, 2.52 mmol) and MnO2 (85% purity, 4.60 g, 45.0 mmol). Eluent: hexane/Et2O = 4 : 1. Spectra data were identical with those of the literature.28)
5a: Orange solid. UV-vis (EtOH) λmax 405 nm (ε = 48000). IR (CHCl3) νmax cm−1: 2926, 1667, 1603, 1559. 1H-NMR (600 MHz, CDCl3) δ: 9.61 (1H, d, J = 7.8 Hz), 7.51 (1H, dd, J = 15.0, 12.0 Hz), 6.92 (1H, dd, J = 14.4, 11.4 Hz), 6.38 (1H, d, J = 15.0 Hz), 6.35 (1H, d, J = 12.0 Hz), 6.29 (1H, d, J = 16.2 Hz), 6.18 (1H, dd, J = 12.6, 7.8 Hz), 6.17 (1H, d, J = 12.6 Hz), 6.15 (1H, d, J = 16.2 Hz), 2.11 (3H, s), 2.04 (2H, t, J = 6.0 Hz), 2.01 (3H, s), 1.72 (3H, s), 1.65–1.58 (2H, m), 1.49–1.45 (2H, m), 1.04 (6H, s). 13C-NMR (150 MHz, CDCl3) δ: 193.56, 147.64, 146.68, 139.45, 137.75, 137.32, 135.51, 130.77, 130.15, 129.96, 129.89, 128.72, 128.48, 39.64, 34.29, 33.15, 28.98, 21.77, 19.22, 13.29, 12.93.
(2E,4E,6E,8E,10E)-5,9-Dimethyl-11-(2,6,6-trimethylcyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaen-1-al (A2-14ex, 5b)According to GP1, ethyl (2E,4E,6E,8E,10E)-5,9-dimethyl-11-(2,6,6-trimethylcyclo-hexa-1,3-dien-1-yl)-2,4,6,8,10-undecapentaenoate (115 mg, 85%) was obtained from 3,4-didehydroretinal22) (2b: 109 mg, 0.387 mmol), triethyl phosphonoacetate (130 mg, 0.581 mmol) and NaH (60%, 23.2 mg, 0.581 mmol). Eluent: hexane/Et2O = 20 : 1. According to GP3, 5b (64.1 mg, 54%, in 3 steps from 2b) was obtained from ethyl (2E,4E,6E,8E,10E)-5,9-dimethyl-11-(2,6,6-trimethyl-cyclohexa-1,3-dien-1-yl)-2,4,6,8,10-undeca-pentaenoate (115 mg, 0.327 mmol), DIBAL (1.31 mL, 1.0 M in hexane, 1.31 mmol) and MnO2 (85% purity,1.15 g, 11.2 mmol). Eluent: hexane/Et2O = 5 : 1. Spectral data were shown in the literature.20)
Preparation of ChR Analogs and SpectroscopyIn this study, two ChR variants, ReaCh (NCBI accession number, KF448069)11) and ChrimsonR (Chrimson (KF992060) K176R mutant)19) were used. These ChR variants were expressed by HEK293 system as reported previously.18) Briefly, the cDNA of these ChR variants were inserted into the mammalian expression vector pCAG GS with the epitope sequence of anti-bovine rhodopsin monoclonal antibody rho1D4 (ETSQVAPA) on the C-terminus. HEK293 cells transfected with the plasmid bearing the ChR gene were collected after incubation for 2 d, and suspended in Buffer P (50 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) and 140 mM NaCl, pH 6.5). Retinal or analog dissolved in ethanol was added to the sample at final concentration of 20 µM. After overnight incubation at 4°C, the regenerated pigments were extracted by 1% n-dodecyl-β-D-maltoside (DDM) in Buffer P, and purified by rho1D4-antibody conjugated agarose column. The final solvent of the sample was Buffer P with 0.02% DDM at pH 6.5. All the procedures above were performed under deep-red light.
UV-visible absorption spectra of pigments were measured by UV-visible spectrophotometer (Shimadzu UV-2400), which is equipped with a water circulation system to the cell holder to keep the sample at 4°C. HEK293 cells produce A1,29) which generates approx. 10% (ReaChR) or approx. 50% (ChrimsonR) of fully regenerated pigment with excess A1. To show the pure spectra of new ChRs, the contribution of ChR regenerated by the endogenous A1 was subtracted from the sample spectra, and the absorption spectra were normalized to absorbance 1.0 at 278 nm.