Regular Articles
Novel Steroidal Glycosides from the Whole Plants of Helleborus foetidus
2020 年 68 巻 3 号 p. 273-287
詳細
2020 年 68 巻 3 号 p. 273-287
Phytochemical analysis of the whole Helleborus foetidus plants identified 28 steroidal glycosides (1–28), including 20 novel spirostanol glycosides (1–20) and a novel furostanol glycoside (21). The structures of the newly identified compounds were elucidated by two-dimensional NMR spectroscopy and hydrolytic cleavage. Compounds 12, 13, and 15 were determined to be spirostanol trisdesmosides bearing sugar moieties at the C-1, -21, and -24 hydroxy groups of the aglycone unit. The isolated compounds were subsequently evaluated for cytotoxic activity against HL-60 human promyelocytic leukemia cells and A549 human lung carcinoma cells. In particular, 7 showed cytotoxic activity against the HL-60 and A549 cells, with IC50 values of 5.9 and 6.6 µM, respectively, whereas 19 was selectively cytotoxic to A549 cells with an IC50 value of 5.5 µM.
Plants from the genus Helleborus are perennials belonging to the Ranunculaceae family, and are widely distributed throughout Europe.1) Helleborus species are poisonous plants and a series of bufadienolides have been isolated from H. caucasicus,2,3) H. odorus,4,5) H. thibetanus,6–8) and H. torquatus.9) Recently, various bufadienolides from H. orientalis and H. foetidus were isolated and identified, some of which showed significantly potent cytotoxic activity and induced apoptosis via a mitochondria dependent pathway in cultured tumor cells.10,11) Because Helleborus species has been reported to concomitantly contain the glycoside derivatives of C27 steroids (spirostanols and furostanols),12–18) further phytochemical examination of the methanolic extract of H. foetidus was performed using whole plants with a specific focus on steroidal glycosides. The isolation of 28 steroidal glycosides (1–28), including 20 novel spirostanol glycosides (1–20) and a novel furostanol glycoside (21) was achieved. The structures of the newly identified compounds were determined from their two-dimensional NMR spectra and hydrolytic cleavage. The cytotoxic activities of the isolated compounds were evaluated against HL-60 human promyelocytic leukemia cells and A549 human lung carcinoma cells.
Whole plants of H. foetidus (fresh weight, 3.3 kg) were extracted using hot MeOH. The concentrated MeOH extract (115 g) was fractionated using a Diaion HP-20 porous polymer polystyrene resin column with MeOH/H2O (3 : 7), MeOH/H2O (1 : 1), MeOH, EtOH, and EtOAc, successively (each solvent volume 6 L). The MeOH eluted fraction was subjected to silica gel column chromatography (CC), octadecylsilanized (ODS) silica gel CC, and preparative ODS HPLC to obtain 28 compounds (1–28). Previously characterized compounds (22–28) were identified as (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-α-L-rhamnopyranosyl-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside (22),19) (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside (23),19) (23S,24S)-3β,23,24-trihydroxyspirosta-5,25(27)-dien-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside (24),20) (23S,24S)-21-acetyloxy-3β,23,24-trihydroxyspirosta-5,25(27)-dien-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside (25),10) (25R)-26-[(β-D-glucopyranosyl)oxy]-1β,22α-dihydroxyfurost-5-en-3β-yl β-D-glucopyranoside (26),21) (25R)-26-[(β-D-glucopyranosyl)oxy]-22α-hydroxyfurost-5-en-3β-yl O-α-L-rhamnopyranosyl-(1→2)-β-D-glucopyranoside (27),22) and (25S)-22α,25-epoxy-26-[(β-D-glucopyranosyl)oxy]-3β-hydroxyfurost-5-en-1β-yl α-L-arabinopyranoside (28),13) respectively (Fig. 1).

Ara: α-L-arabinopyranosyl; Api: β-D-apiofuranosyl; Fuc: β-D-fucopyranosyl; Gal: β-D-galactopyranosyl; Glc: β-D-glucopyranosyl; Rha: α-L-rhamnopyranosyl; Qui: β-D-quinovopyranosyl; Ac: acetyl.
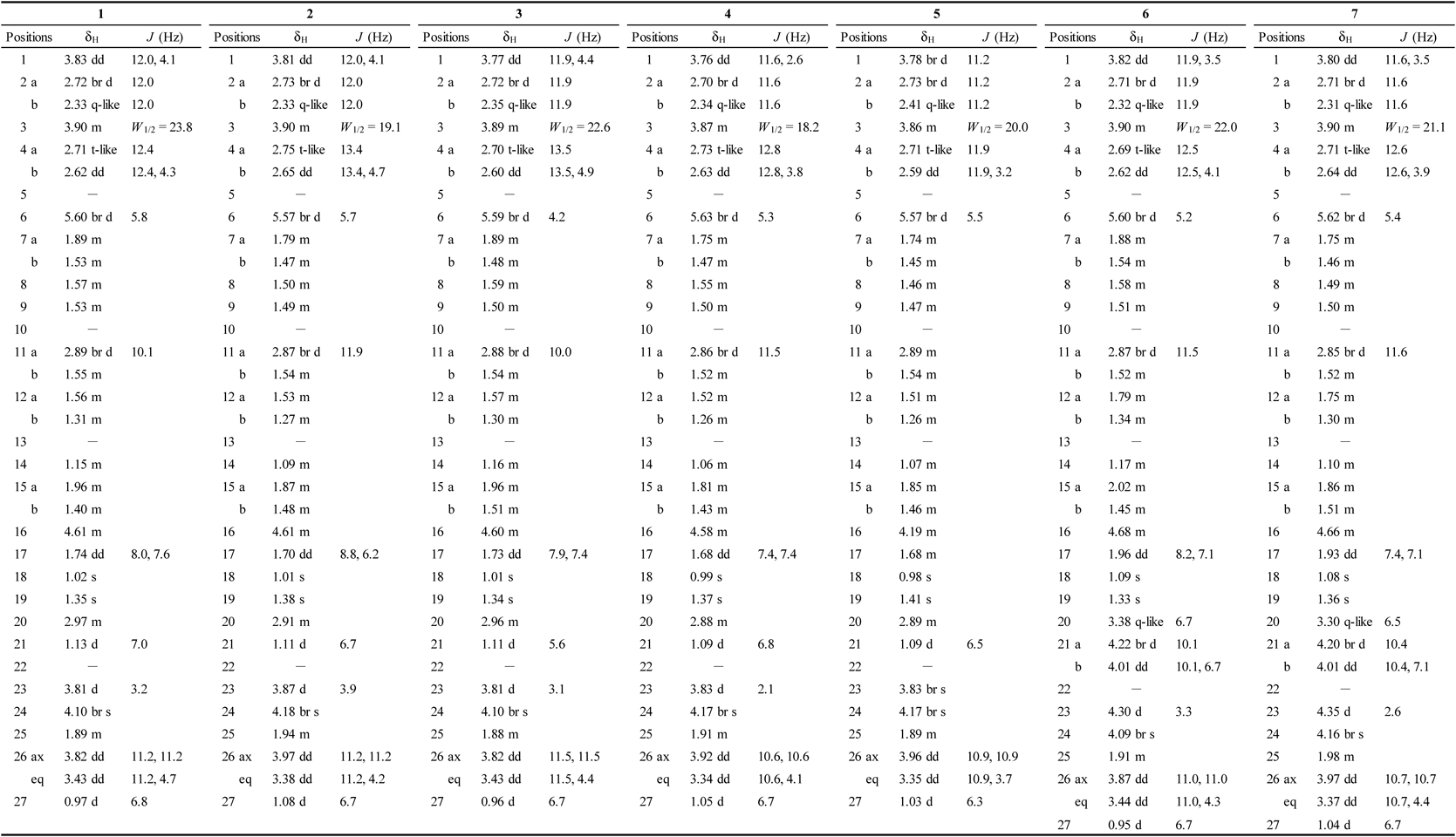
Compound 1 was obtained as an amorphous powder and its molecular formula was determined to be C45H70O19, based on high resolution-electrospray ionization-time of flight (HR-ESI-TOF)-MS and 13C-NMR spectra data. The IR spectrum of 1 showed absorption bands of hydroxy (3392 cm−1) and carbonyl groups (1729 cm−1). The 1H- and 13C-NMR spectra of 1 indicated the presence of four steroidal methyl groups [δH 1.35 (3H, s, Me-19), 1.13 (3H, d, J = 7.0 Hz, Me-21), 1.02 (3H, s, Me-18), and 0.97 (3H, d, J = 6.8 Hz, Me-27); δC 16.8 (C-18), 14.9 (C-19), 14.6 (C-21), and 13.0 (C-27)], an olefinic group [δH 5.60 (1H, br d, J = 5.8 Hz, H-6); δC 139.3 (C-5) and 124.9 (C-6)], an oxygenated methylene group [δH 3.82 (1H, dd, J = 11.2, 11.2 Hz, H-26ax) and 3.43 (1H, dd, J = 11.2, 4.7 Hz, H-26eq); δC 60.6 (C-26)], five oxygenated methine groups [δH 4.61 (1H, m, H-16), 4.10 (1H, br s, H-24), 3.90 (1H, m, H-3), 3.83 (1H, dd, J = 12.0, 4.1 Hz, H-1), and 3.81 (1H, d, J = 3.2 Hz, H-23); δC 84.0 (C-1), 83.0 (C-16), 73.0 (C-24), 68.7 (C-23), and 68.0 (C-3)], and an acetyl group [δH 2.01 (3H, s); δC 170.8 and 21.0]. Furthermore, signals corresponding to three anomeric protons and carbons were observed [δH 6.48 (1H, br s), 4.94 (1H, d, J = 7.6 Hz), and 4.69 (1H, d, J = 7.9 Hz); δC 106.7, 100.8, and 100.7]. The 1H- and 13C-NMR spectral data of 1 were similar to those obtained for 24, except for the signals arising from the F-ring part of the steroidal skeleton. Instead of the 1H- and 13C-NMR signals arising from the C-25(27) exomethylene group [δH 5.07 and 4.97 (each 1H, br s, H2-27); δC 146.1 (C-25) and 112.3 (C-27)] observed in 24, signals from a secondary methyl group [δH 0.97 (3H, d, J = 6.8 Hz, Me-27); δC 13.0 (C-27)] and methine group [δH 1.89 (1H, m, H-25); δC 36.1 (C-25)] were observed in the 1H- and 13C-NMR spectra of 1. These data suggest that 1 is a saturated derivative of 24 in terms of the C-25(27) exomethylene group. Nuclear Overhauser effect spectroscopy (NOESY) of 1 was performed and NOE correlations were observed between H-23 (δH 3.81) and H-20 (δH 2.97)/H-24 (δH 4.10)/H-25 (δH 1.89) as well as between H-26ax (δH 3.82) and H-16 (δH 4.61). Additionally, the proton spin-coupling constants of 3JH-23,H-24 and 3JH-25,H-26ax were 3.2 and 11.2 Hz, respectively. Thus, the configurations of C-23, C-24, and C-25 were determined as 23S, 24S, and 25S. Acid hydrolysis of 1 with 0.2 M HCl (dioxane/H2O; 1 : 1) yielded L-arabinose, L-rhamnose, and D-xylose carbohydrate moieties. The 1H-1H correlation spectroscopy (COSY) and heteronuclear single quantum coherence (HSQC) spectra indicated that the sugar moiety of 1 comprised a 2,3-disubstituted inner α-L-arabinopyranosyl unit [Ara: δH 4.69 (1H, d, J = 7.9 Hz, H-1′); δC 100.8, 72.8, 85.2, 69.9, and 67.3 (C-1′–5′)], a terminal α-L-rhamnopyranosyl unit [Rha: δH 6.48 (1H, br s, H-1″); δC 100.7, 72.2, 69.9, 76.5, 66.5, and 18.5 (C-1″–6″)], and a terminal β-D-xylopyranosyl unit [Xyl: δH 4.94 (1H, d, J = 7.6 Hz, H-1‴); δC 106.7, 74.5, 78.5, 70.9, and 67.1 (C-1‴–5‴)]. In the heteronuclear multiple bond correlation (HMBC) spectrum of 1, long-range correlations were observed between H-1″ of Rha (δH 6.48) and C-2′ of Ara (δC 72.8), H-1‴ of Xyl (δH 4.94) and C-3′ of Ara (δC 85.2), between H-1′ of Ara (δH 4.69) and C-1 of the aglycone (δC 84.0). Furthermore, a 3JC,H correlation was observed between H-4″ of Rha (δH 5.78) and the acetyl carbonyl carbon (δC 170.8). Accordingly, 1 was determined to be (23S,24S,25S)-3β,23,24-trihydroxyspirost-5-en-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
The 1H- and 13C-NMR spectra of 2 (C51H80O23) indicated that 2 was analogous to 1, including the triglycoside moiety attached to C-1 of the aglycone. However, the molecular formula of 2 was larger than that of 1 by a C6H10O4 unit, indicating the presence of an additional hexosyl unit. Acid hydrolysis of 2 yielded L-arabinose, L-rhamnose, D-quinovose, and D-xylose, while 1H-1H COSY and HSQC analysis indicated that 2 contained a terminal β-D-quinovopyranosyl unit [Qui: δH 5.24 (1H, d, J = 7.6 Hz, H-1″″); δC 105.3, 76.2, 78.1, 76.9, 73.0, and 18.6 (C-1″″–6″″)]. Comparing the 13C-NMR spectrum of 2 with that of 1, the signal attributed to C-24 of the aglycone was shifted downfield by 8.4 ppm. Additionally, the HMBC spectrum of 2 showed a long-range correlation between H-1″″ of Qui (δH 5.24) and C-24 of the aglycone (δC 81.4). Therefore, 2 was determined to be (23S,24S,25S)-24-[(β-D-quinovopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
Comparison of the 1H- and 13C-NMR spectra of 3 (C50H78O23) with those of 1 suggested that 3 contained the same aglycone moiety as 1 but differed in terms of the sugar moiety attached to C-1 of the aglycone. Acid hydrolysis of 3 afforded L-arabinose, D-apiose, L-rhamnose, and D-xylose, while 1H-1H COSY and HSQC analysis of the sugar moiety in 3 showed that it consisted of a 2,3-disubstituted α-L-arabinopyranosyl (Ara), 3,4-disubstituted α-L-rhamnopyranosyl (Rha), terminal β-D-xylopyranosyl (Xyl), and terminal β-D-apiofuranosyl units [Api: δH 5.95 (1H, br s, H-1″″); δC 112.1, 77.8, 80.0, 74.8, and 65.2 (C-1″″–5″″)], as well as an acetyl group. The α-anomeric configuration of Rha was determined by the large 1JC-1,H-1 value of 169.7 Hz. In the HMBC spectrum of 3, long-range correlations were observed between H-1″″ of Api (δH 5.95) and C-3″ of Rha (δC 77.7), H-1″ of Rha (δH 6.49) and C-2′ of Ara (δC 72.4), H-1‴ of Xyl (δH 4.92) and C-3′ of Ara (δC 85.2), H-4″ of Rha (δH 5.90) and the acetyl carbonyl carbon (δC 170.7), and between H-1′ of Ara (δH 4.63) and C-1 of the aglycone (δC 84.2). Thus, 3 was determined to be (23S,24S,25S)-3β,23,24-trihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
Compound 4 (C56H88O27) exhibited 1H- and 13C-NMR spectral features similar to those of 3, except for the signals arising from the F-ring part of the steroidal skeleton. The molecular formula of 4 featured an additional C6H10O4 compared to that of 3. From acid hydrolysis of 4, L-arabinose, D-apiose, D-fucose, L-rhamnose, and D-xylose were obtained. A terminal β-D-fucopyranosyl unit [Fuc: δH 5.13 (1H, d, J = 7.9 Hz, H-1″‴); δC 105.8, 73.1, 75.1, 72.6, 71.3, and 17.1 (C-1″‴–6″‴)] was detected by 1H-1H COSY and HSQC spectral analysis of 4. Comparing the 13C-NMR spectrum of 4 with that of 3, the signal assigned to C-24 of the aglycone was shifted downfield by 8.4 ppm and a 3JC,H correlation was observed between H-1″‴ of Fuc (δH 5.13) and C-24 of the aglycone (δC 81.4) in the HMBC spectrum. Therefore, 4 was elucidated as (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
The 1H- and 13C-NMR spectra of 5 (C49H78O23) were essentially analogous to those of 22, but remarkable differences were observed for the signals assigned to the monosaccharide linked to C-24 of the aglycone between the two glycosides. Acid hydrolysis of 5 yielded L-arabinose, D-glucose, L-rhamnose, and D-xylose. Analysis of the associated 1H-1H COSY and HSQC spectra indicated the presence of a terminal β-D-glucopyranosyl unit [Glc: δH 5.33 (1H, d, J = 7.7 Hz, H-1″″); δC 105.6, 76.1, 78.3, 69.5, 78.3, and 63.0 (C-1″″–6″″)]. The HMBC spectrum of 5 showed a long-range correlation between H-1″″ of Glc (δH 5.33) and C-24 of the aglycone (δC 81.6). Accordingly, 5 was determined to be (23S,24S,25S)-24-[(β-D-glucopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-α-L-rhamnopyranosyl-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
The 1H- and 13C-NMR spectra of 6 (C45H70O20), 7 (C51H80O24), and 8 (C50H78O24) were closely related to those of 1, 23, and 3, respectively. The molecular formula of 6 was larger than that of 1 (C45H70O19) by a single oxygen atom, and only differences observed in the 1H- and 13C-NMR signals were those assigned to Me-21/C-21. The methyl signals [δH 1.13 (3H, d, J = 7.0 Hz); δC 14.6] in 1 were displaced by hydroxymethyl signals [δH 4.22 (1H, br d, J = 10.1 Hz) and 4.01 (1H, dd, J = 10.1, 6.7 Hz); δC 62.3] in 6. Similarly, the molecular formulas of 7 and 8 were larger than those of 23 (C51H80O23) and 3 (C50H78O23) by a single oxygen atom, and the C-21 methyl groups were hydroxylated, as indicated by the 1H- and 13C-NMR spectra [7: δH 4.20 (1H, br d, J = 10.4 Hz) and 4.01 (1H, dd, J = 10.4, 7.1 Hz); δC 62.5; 8: δH 4.19 (1H, m), 3.98 (1H, m); δC 62.3)]. Therefore, 6, 7, and 8 were identified as the corresponding C-21 hydroxy derivatives of 1, 23, and 3, respectively, establishing them as (23S,24S,25S)-3β,21,23,24-tetrahydroxyspirost-5-en-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-3β,21,23-trihydroxyspirost-5-en-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, and (23S,24S,25S)-3β,21,23,24-tetrahydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, respectively.
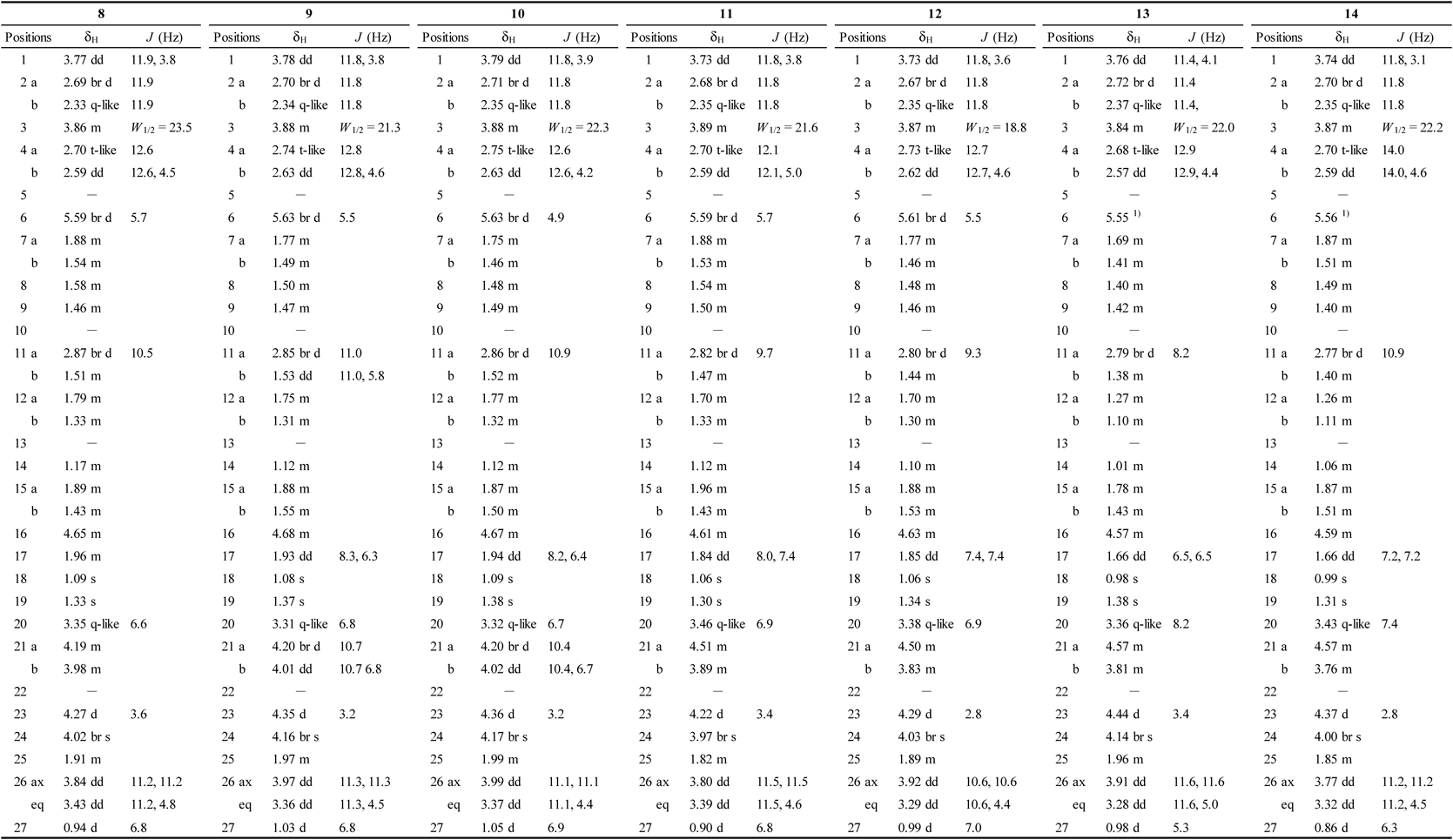
The 1H- and 13C-NMR spectra of 9 (C55H86O28) were similar to those obtained for 8, except for the presence of the signals attributed to one more monosaccharide unit. The molecular formula of 9 was larger than that of 8 by C5H8O4, corresponding to a single pentosyl unit. Acid hydrolysis of 9 yielded L-arabinose, D-apiose, L-rhamnose, and D-xylose. Analysis of the corresponding 1H-1H COSY and HSQC spectra of the sugar moieties of 9 indicated the presence of an additional terminal α-L-arabinopyranosyl unit [Ara′: δH 5.11 (1H, d, J = 7.7 Hz, H-1″‴); δC 106.4, 73.6, 74.8, 69.9, and 67.2 (C-1″‴–5″‴)]. A long-range correlation was observed between H-1″‴ of Ara′ (δH 5.11) and C-24 of the aglycone (δC 81.7) in the HMBC spectrum. Thus, 9 was identified as (23S,24S,25S)-24-[(β-D-arabinopyranosyl)oxy]-3β,21,23-trihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
Analysis of the 1H- and 13C-NMR spectra of 10 (C56H88O28) and subsequent comparison with those of 9 revealed that the aglycone and tetraglycoside attached to C-1 of the aglycone were identical to those of 9 but differed in terms of the monosaccharide linked to C-24 of the aglycone. Instead of the signals for an arabinopyranosyl moiety, signals attributed to a β-D-fucopyranosyl (Fuc) residue were observed. Acid hydrolysis of 10 yielded L-arabinose, D-apiose, D-fucose, L-rhamnose, and D-xylose. The linkage of the Fuc moiety to C-24 of the aglycone was confirmed by an HMBC correlation between H-1″‴ of Fuc (δH 5.13) and C-24 of the aglycone (δC 81.7). Thus, 10 was assigned as (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-3β,21,23-trihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
The 1H- and 13C-NMR spectra of 11 (C56H88O29) suggested that it was analogous to 8, including the tetraglycoside moiety attached to C-1 of the aglycone. The molecular formula of 11 was larger than that of 8 by C6H10O5, corresponding to a single hexosyl unit. Acid hydrolysis of 11 yielded L-arabinose, D-apiose, D-galactose, L-rhamnose, and D-xylose. The 1H-1H COSY and HSQC spectral analysis of 11 indicated the presence of a terminal β-D-galactopyranosyl unit [Gal: δH 4.90 (1H, d, J = 7.9 Hz, H-1″‴); δC 105.4, 72.5, 75.6, 70.2, 77.0, and 62.4 (C-1″‴–6″‴)], and an HMBC correlation was observed between H-1″‴ of Gal (δH 4.90) and C-21 of the aglycone (δC 70.0). Therefore, 11 was determined to be (23S,24S,25S)-21-[(β-D-galactopyranosyl)oxy]-3β,23,24-trihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
The 1H- and 13C-NMR spectra of 12 (C61H96O33) were similar to those obtained for 11, but the molecular formula of 12 was larger than that of 11 by C5H8O4. Acid hydrolysis of 12 afforded L-arabinose, D-apiose, D-galactose, L-rhamnose, and D-xylose. The 1H-1H COSY and HSQC spectral analysis of 12 indicated the presence of signals corresponding to an additional terminal α-L-arabinopyranosyl (Ara′) unit. The HMBC spectrum of 12 contained a long-range correlation from H-1‴‴ of Ara′ (δH 5.10) to C-24 of the aglycone (δC 81.4). Thus, 12 was assigned as (23S,24S,25S)-24-[(β-D-arabinopyranosyl)oxy]-21-[(β-D-galactopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
Compound 13 (C61H98O33) was obtained as an amorphous powder and its 1H- and 13C-NMR spectra showed the same aglycone unit as 7, also indicating that the sugar moieties attached to C-1 and C-24 of the aglycone of 13 were coincident with those of 22. Additionally, a 2-substituted β-D-galactopyranosyl unit [Gal: δH 4.98 (1H, d, J = 7.4 Hz, H-1″″); δC 103.4, 81.6, 75.2, 69.7, 77.0, and 62.2 (C-1″″–6″″)] and a terminal β-D-glucopyranosyl unit [Glc: δH 5.40 (1H, d, J = 7.6 Hz, H-1″‴); δC 106.1, 76.5, 78.1, 71.4, 78.6, and 62.5 (C-1″‴–6″‴)] were detected in the 1H-1H COSY and HSQC spectral analysis of 13. Acid hydrolysis of 13 afforded L-arabinose, D-fucose, D-galactose, D-glucose, L-rhamnose, and D-xylose. In the HMBC spectrum of 13, long-range correlations were observed between H-1″‴ of Glc (δH 5.40) and C-2″″ of Gal (δC 81.6) as well as between H-1″″ of Gal (δH 4.98) and C-21 of the aglycone (δC 69.9). Thus, 13 was identified as (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-21-[O-β-D-glucopyranosyl-(1→2)-(β-D-galactopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-α-L-rhamnopyranosyl-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
The 1H- and 13C-NMR spectra of 14 (C62H98O34) and 15 (C68H108O38) indicated their relation to 8 and 10, respectively, containing a diglycoside composed of Glc and Gal at C-21 of the aglycone, as in 13. The molecular formula of 14 and 15 were both larger than those of 8 and 10, respectively, by C12H20O10 and acid hydrolysis of 14 yielded L-arabinose, D-galactose, D-glucose, L-rhamnose, and D-xylose. In contrast, acid hydrolysis of 15 afforded L-arabinose, D-fucose, D-galactose, D-glucose, L-rhamnose, and D-xylose. In the HMBC spectra of 14 and 15, long-range correlations were observed between H-1‴‴ of Glc [δH 5.38 (14); 5.38 (15)] and C-2″‴ of Gal [δC 81.6 (14); 81.8 (15)] as well as between H-1″‴ of Gal [δH 4.98 (14); 4.97 (15)] and C-21 of the aglycone [δC 69.8 (14); 69.8 (15)]. Accordingly, 14 and 15 were identified as (23S,24S,25S)-21-[O-(β-D-glucopyranosyl-(1→2)-β-D-galactopyranosyl)oxy]-3β,23,24-trihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside and (23S,24S,25S)-24-[(β-D-fucopyranosyl)oxy]-21-[O-(β-D-glucopyranosyl-(1→2)-β-D-galactopyranosyl)oxy]-3β,23-dihydroxyspirost-5-en-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, respectively.
The NMR spectral features of 16 (C56H86O27), 17 (C50H76O24), and 18 (C55H84O28) were related to those of 4, 8, and 9, respectively. However, instead of containing three-proton doublet signals for Me-27, as observed in the 1H- and 13C-NMR spectra of 4, 8, and 9, signals arising from an exomethylene group [δH 5.23 (br s) and 5.08 (br s) (16); 5.05 (br s) and 4.95 (br s) (17); 5.20 (br s) and 5.06 (br s) (18); δC 113.7 (16); 112.3 (17); 113.9 (18)] and quaternary carbon [δC 143.8 (16); 146.2 (17); 143.7 (18)] were observed. Therefore, 16, 17, and 18 correspond to the C-25/27 dehydroxy derivatives of 4, 8, and 9, respectively, as additionally confirmed by long-range correlations from H2-27 to C-24, C-25, and C-26 in the associated HMBC spectra. Therefore, 16, 17, and 18 were identified as (23S,24S)-24-[(β-D-fucopyranosyl)oxy]-3β,23-dihydroxyspirosta-5,25(27)-dien-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, (23S,24S)-3β,21,23,24-tetrahydroxyspirosta-5,25(27)-dien-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, and (23S,24S)-24-[(α-L-arabinopyranosyl)oxy]-3β,21,23-trihydroxyspirosta-5,25(27)-dien-1β-yl O-β-D-apiofuranosyl-(1→3)-O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside, respectively.
Comparison of the 1H- and 13C-NMR spectra 19 (C57H86O29) with those of 18 showed considerable structural similarity. Furthermore, the presence of an additional acetyl group to that attached at C-21 of the aglycone was indicated by the 1H- and 13C-NMR spectra [δH 1.92 (3H, s); δC 170.8 and 20.9]. The ester linkage at aglycone C-21 was formed from acetic acid, as indicated by the long-range correlations between the H2-21 methylene protons [δH 4.37 (m) and 4.34 (m)] and acetyl carbonyl carbon (δC 170.8). Thus, 19 was identified as (23S,24S)-21-acetyloxy-24-[(α-L-arabinopyranosyl)oxy]-3β,23-dihydroxyspirosta-5,25(27)-dien-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
Analysis of the 1H- and 13C-NMR spectra of 20 (C52H78O25) and comparison with those of 19 showed that the aglycone with an acetyloxy group at C-21 and monosaccharide at C-24 were identical to those observed in 19. However, the 1H- and 13C-NMR signals for the apiofuranosyl group linked to C-3 of the rhamnopyranosyl unit in the sugar moiety attached to the aglycone C-1 were not observed. The 1H- and 13C-NMR spectra of the triglycoside moiety agreed with those observed in the concomitantly isolated spirostanol triglycosides 1, 2, 6, and 7. Thus, 20 was identified as (23S,24S)-21-acetyloxy-24-[(α-L-arabinopyranosyl)oxy]-3β,23-dihydroxyspirosta-5,25(27)-dien-1β-yl O-(4-O-acetyl-α-L-rhamnopyranosyl)-(1→2)-O-[β-D-xylopyranosyl-(1→3)]-α-L-arabinopyranoside.
Compound 21 was obtained as an amorphous powder with a molecular formula was of C38H62O14 as determined by HR-ESI-TOF-MS and 13C-NMR data. In the 1H- and 13C-NMR spectra of 21, signals corresponding to four steroidal methyl groups [δH 1.27 (3H, d, J = 7.1 Hz, Me-21), 1.21 (3H, s, Me-19), 0.95 (3H, d, J = 6.5 Hz, Me-27), and 0.90 (3H, s, M-18); δC 17.4 (C-27), 16.7 (C-18), 16.3 (C-21), and 14.7 (C-19)], an olefinic group [δH 5.55 (1H, br d, J = 4.6 Hz, H-6); δC 139.5 (C-5) and 124.7 (C-6)], two anomeric protons and associated carbons [δH 4.79 (1H, d, J = 7.7 Hz) and 4.75 (1H, d, J = 7.1 Hz); δC 104.8 and 102.2], and a hemiacetal carbon [δC 110.6 (C-22)] were observed. These data suggested that 21 was a 22-hydroxyfurostanol glycoside and its enzymatic hydrolysis with naringinase yielded (25R)-3β-hydroxyspirost-5-en-1β-yl α-L-arabinopyranoside (21a)22) and D-glucose. In the associated HMBC spectrum, a long-range correlation was observed between H-1″ of the β-D-glucopyranosyl unit (δH 4.79) and C-26 of the aglycone (δC 75.2). Accordingly, 21 was identified as (25R)-26-[(β-D-glucopyranosyl)oxy]-3β,22α-dihydroxyfurost-5-en-1β-yl α-L-arabinopyranoside.
Compounds 12, 13, and 15 are spirostanol trisdesmosides, which are types of steroidal glycosides rarely observed in the plant kingdom.
The cytotoxic activities of 1–28 were evaluated against HL-60 and A549 cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay method. Compound 7 showed cytotoxic activity against HL-60 and A549 cells with IC50 values of 5.9 ± 0.46 and 6.6 ± 0.17 µM, respectively. In contrast, 19 was selectively cytotoxic against only A549 cells with an IC50 value of 5.5 ± 0.61 µM. Cisplatin was used as a positive control, yielding IC50 values of 1.9 and 3.7 µM for HL-60 and A549 cells, respectively. Compounds 1–6, 8–18, and 20–28 did not show cytotoxicity against HL-60 and A549 cells at concentrations of up to 10 µM.
Optical rotations were measured on a JASCO P-1030 automatic digital polarimeter (Jasco, Tokyo, Japan). IR spectra were obtained using a JASCO FT-IR 620 spectrophotometer (Jasco). NMR spectra were recorded on a Bruker DRX-500 or AV-600 spectrometer (Bruker, Karlsruhe, Germany) using standard Bruker pulse programs. Chemical shifts (δ) were given with reference to tetramethylsilane (TMS) as an internal standard. HR-ESI-TOF-MS data were obtained using a Water-Micromass LCT mass spectrometer (Waters-Micromass, Manchester, U.K.). CC was conducted by Diaion HP-20 (Mitsubishi-chemical, Tokyo, Japan), silica gel Chromatorex BW-300 (Fuji-Silysia Chemical, Aichi, Japan), and ODS silica gel COSMOSIL 75C18-OPN (Nacalai Tesque, Kyoto, Japan). TLC was carried out precoated silica gel 60 F254 or RP18 F254S plates (0.25 mm thick) (Merck, Darmstadt, Germany), and the spots were detected by spraying the plates with 10% H2SO4 aqueous solution and then heating. HPLC was conducted using a system consisting of a CCPM pump (Shimadzu, Kyoto, Japan), an RI-8021 (Tosoh, Tokyo, Japan) or a Shodex OR-2 (Showa-Denko, Tokyo, Japan) detector, and a Rheodyne injection port (Rohnert Park, CA, U.S.A.). A TSK gel ODS-100Z column (10 mm i.d. × 250 mm, 5 µm) (Tosoh) was used for the preparative HPLC. Purities of the isolated compounds were verified by MS, TLC, and NMR spectra. The following materials and biochemical grade reagents were used for the cell cultures and cytotoxicity assays: SH-1300 Lab microplate reader (CORONA ELECTRIC, Ibaraki, Japan); 96-well flat-bottom plate (Iwaki Glass, Chiba, Japan); fetal bovine serum (FBS), 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) solution, RPMI-1640 medium, minimum essential medium (MEM), cisplatin, and MTT (Sigma, St. Louis, MO, U.S.A.); paraformaldehyde and phosphate-buffered saline (PBS) (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan); penicillin G sodium salt and streptomycin sulfate (Gibco, Gland Island, NY, U.S.A.); HL-60 cells (JCRB0085) and A549 cells (JCRB0076) (Human Science Research Resources Bank, Osaka, Japan).
Plant MaterialThe whole plants of H. foetidus were purchased from Fuji-engei (Okayama, Japan). A voucher specimen was deposited at the herbarium of the Tokyo University of Pharmacy and Life Sciences (KS-2014-011).23)
Extraction and IsolationThe whole plants of H. foetidus (fresh weight, 3.3 kg) were extracted with hot MeOH (10 L). After solvent was removed in vacuo, the concentrated MeOH extract (115 g) was fractionated by Diaion HP-20 column and then successively eluted with MeOH/H2O (3 : 7), MeOH/H2O (1 : 1), MeOH, EtOH, and EtOAc (each solvent volume 6 L). The MeOH eluate fraction (10 g) was chromatographed on silica gel eluted with stepwise gradient of CHCl3/MeOH/H2O (90 : 10 : 1, 40 : 10 : 1, 20 : 10 : 1) and finally MeOH alone to get 17 fractions (Frs. 1–17). Fraction 10 was subjected to silica gel CC eluted with CHCl3/MeOH/H2O (90 : 10 : 1; 40 : 10 : 1) and EtOAc/MeOH/H2O (80 : 10 : 1; 50 : 10 : 1; 40 : 10 : 1), and ODS silica gel CC eluted with MeOH/H2O (3 : 2; 1 : 1) and MeCN/H2O (7 : 3; 1 : 1; 3 : 7; 1 : 3) to yield 1 (35.7 mg), 21 (85.4 mg), 23 (72.1 mg), and 24 (2.9 mg). Fraction 11 was separated by ODS silica gel CC using MeOH/H2O (4 : 1; 3 : 2) and MeCN/H2O (1 : 3) to obtain 2 (5.8 mg), 3 (27.0 mg), 6 (3.3 mg), 25 (18.1 mg), and 28 (6.7 mg). Fraction 13 was chromatographed on silica gel eluted with CHCl3/MeOH/H2O (30 : 10 : 1), ODS silica gel eluted with MeOH/H2O (3 : 2) and MeCN/H2O (1 : 3; 1 : 4), and preparative HPLC using MeCN/H2O (1 : 2; 3 : 7) to furnish 4 (72.1 mg), 5 (2.3 mg), 7 (30.1 mg), 8 (13.0 mg), 20 (8.5 mg), and 26 (85.4 mg). Fraction 14 was fractionated by ODS silica gel CC using MeOH/H2O (3 : 2; 11 : 9; 1 : 1) and MeCN/H2O (1 : 3; 1 : 4), and preparative HPLC using MeCN/H2O (3 : 7; 1 : 3) to acquire 16 (16.2 mg), 17 (6.1 mg), 19 (28.6 mg), 22 (16.6 mg), and 27 (4.9 mg). Fraction 15 was separated by ODS silica gel CC eluted with MeOH/H2O (11 : 9; 1 : 1) and MeCN/H2O (1 : 3; 1 : 4), and preparative HPLC using MeCN/H2O (1 : 3) to afford 9 (26.9 mg) and 10 (6.8 mg). Fraction 16 was chromatographed on ODS silica gel using MeOH/H2O (11 : 9; 9 : 11) and MeCN/H2O (1 : 3; 1 : 4) to collect 11 (3.9 mg) and 18 (46.3 mg). Fraction 17 was subjected to silica gel CC eluted with CHCl3/MeOH/H2O (20 : 10 : 1; 10 : 10 : 1), ODS silica gel CC eluted with MeOH/H2O (1 : 1) and MeCN/H2O (1 : 3; 1 : 4), and preparative HPLC using MeCN/H2O (1 : 3; 1 : 4) to furnish 12 (12.9 mg), 13 (7.0 mg), 14 (19.9 mg), and 15 (15.9 mg).
Compound 1Amorphous powder; [α]D25 −94.4 (c = 0.10, MeOH); IR νmax (film) cm−1: 3392 (OH), 2926 (CH), 1729 (C=O); 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 937.4412 [M + Na]+ (Calcd for C45H70NaO19: 937.4409).
 |
 |
 |
All spectra were measured in C5D5N (1, 8, 18, and 19: 500 MHz; 2-7, 9-17, 20, and 21: 600 MHz). 1) Signals are unclear due to overlapping with water signal.
 |
All spectra were measured in C5D5N (1, 8, 18, and 19: 125 MHz; 2–7, 9–17, 20, and 21: 150 MHz).
 |
 |
 |
All spectra were measured in C5D5N (1, 8, 18, and 19: 500 MHz; 2–7, 9–17, 20, and 21: 600 MHz).
 |
All spectra were measured in C5D5N (1, 8, 18, and 19: 125 MHz; 2–7, 9–17, 20, and 21: 150 MHz).
Amorphous powder; [α]D25 −14.5 (c = 0.05, MeOH); IR νmax (film) cm−1: 3370 (OH), 2925 (CH), 1730 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1083.4980 [M + Na]+ (Calcd for C51H80NaO23: 1083.4988).
Compound 3Amorphous powder; [α]D25 −23.4 (c = 0.10, MeOH); IR νmax (film) cm−1: 3375 (OH), 2930 (CH), 1727 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1069.4832 [M + Na]+ (Calcd for C50H78NaO23: 1069.4832).
Compound 4Amorphous powder; [α]D25 −31.7 (c = 0.11, MeOH); IR νmax (film) cm−1: 3375 (OH), 2927 (CH), 1727 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1215.5405 [M + Na]+ (Calcd for C56H88NaO27: 1215.5411).
Compound 5Amorphous powder; [α]D25 −8.7 (c = 0.14, MeOH); IR νmax (film) cm−1: 3374 (OH), 2925 (CH); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1057.4819 [M + Na]+ (Calcd for C49H78NaO23: 1057.4832).
Compound 6Amorphous powder; [α]D25 −30.5 (c = 0.05, MeOH); IR νmax (film) cm−1: 3364 (OH), 2925 (CH), 1725 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 953.4354 [M + Na]+ (Calcd for C45H70NaO20: 953.4358).
Compound 7Amorphous powder; [α]D25 −42.0 (c = 0.10, MeOH); IR νmax (film) cm−1: 3383 (OH), 2922 (CH), 1728 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1099.4934 [M + Na]+ (Calcd for C51H80NaO24: 1099.4937).
Compound 8Amorphous powder; [α]D25 −13.9 (c = 0.10, MeOH); IR νmax (film) cm−1: 3389 (OH), 2924 (CH), 1729 (C=O); 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1085.4775 [M + Na]+ (Calcd for C50H78NaO24: 1085.4781).
Compound 9Amorphous powder; [α]D25 −62.5 (c = 0.10, MeOH); IR νmax (film) cm−1: 3388 (OH), 2925 (CH), 1731 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1217.5203 [M + Na]+ (Calcd for C55H86NaO28: 1217.5203).
Compound 10Amorphous powder; [α]D25 −41.9 (c = 0.05, MeOH); IR νmax (film) cm−1: 3389 (OH), 2925 (CH), 1731 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1231.5364 [M + Na]+ (Calcd for C56H88NaO28: 1231.5360).
Compound 11Amorphous powder; [α]D25 −8.8 (c = 0.05, MeOH); IR νmax (film) cm−1: 3357 (OH), 2926 (CH), 1731 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1247.5299 [M + Na]+ (Calcd for C56H88NaO29: 1247.5309).
Compound 12Amorphous powder; [α]D25 −22.7 (c = 0.10, MeOH); IR νmax (film) cm−1: 3357 (OH), 2930 (CH), 1732 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1379.5753 [M + Na]+ (Calcd for C61H96NaO33: 1379.5732).
Compound 13Amorphous powder; [α]D25 −17.7 (c = 0.10, MeOH); IR νmax (film) cm−1: 3364 (OH), 2927 (CH); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1381.5883 [M + Na]+ (Calcd for C61H98NaO33: 1381.5888).
Compound 14Amorphous powder; [α]D25 −34.1 (c = 0.10, MeOH); IR νmax (film) cm−1: 3360 (OH), 2925 (CH), 1730 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1409.5863 [M + Na]+ (Calcd for C62H98NaO34: 1409.5837).
Compound 15Amorphous powder; [α]D25 −32.0 (c = 0.10, MeOH); IR νmax (film) cm−1: 3373 (OH), 2931 (CH), 1731 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1555.6422 [M + Na]+ (Calcd for C68H108NaO38: 1555.6416).
Compound 16Amorphous powder; [α]D25 −69.1 (c = 0.10, MeOH); IR νmax (film) cm−1: 3389 (OH), 2932 (CH), 1732 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1213.5251 [M + Na]+ (Calcd for C56H86NaO27: 1213.5254).
Compound 17Amorphous powder; [α]D25 −94.4 (c = 0.05, MeOH); IR νmax (film) cm−1: 3375 (OH), 2925 (CH), 1731 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1083.4623 [M + Na]+ (Calcd for C50H76NaO24: 1083.4624).
Compound 18Amorphous powder; [α]D25 −125.4 (c = 0.10, MeOH); IR νmax (film) cm−1: 3404 (OH), 2923 (CH), 1728 (C=O); 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1215.5081 [M + Na]+ (Calcd for C55H84NaO28: 1215.5047).
Compound 19Amorphous powder; [α]D25 −110.3 (c = 0.10, MeOH); IR νmax (film) cm−1: 3393 (OH), 2924 (CH), 1733 (C=O); 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (500 MHz, C5D5N) and 13C-NMR (125 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1257.5111 [M + Na]+ (Calcd for C57H86NaO29: 1257.5152).
Compound 20Amorphous powder; [α]D25 −43.6 (c = 0.025, MeOH); IR νmax (film) cm−1: 3406 (OH), 2923 (CH), 1720 (C=O); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar and acetyl moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 1125.4730 [M + Na]+ (Calcd for C52H78NaO25: 1125.4730).
Compound 21Amorphous powder; [α]D25 −10.7 (c = 0.11, MeOH); IR νmax (film) cm−1: 3364 (OH), 2925 (CH); 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the aglycone moiety, see Tables 1, 2; 1H- (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data of the sugar moieties, see Tables 3, 4; HR-ESI-TOF-MS m/z: 765.4035 [M + Na]+ (Calcd for C38H62NaO14: 765.4037).
Acid Hydrolysis of 1–20Compound 1 (10.0 mg), 2 (2.0 mg), 3 (3.6 mg), 4 (4.0 mg), 5 (1.9 mg), 6 (1.1 mg), 7 (10.0 mg), 8 (5.0 mg), 9 (5.5 mg), 10 (2.3 mg), 11 (2.5 mg), 12 (4.7 mg), 13 (4.0 mg), 14 (5.0 mg), 15 (5.0 mg), 16 (4.8 mg), 17 (2.4 mg), 18 (10.0 mg), 19 (5.0 mg), and 20 (2.8 mg) were independently dissolved in 0.2 M HCl or 0.5 M HCl (dioxane/H2O, 1 : 1) and heated at 95°C for 30 min under an Ar atmosphere. Each reaction mixture was neutralized by passing through an Amberlite IRA-96 (Organo, Tokyo, Japan) column, was subjected to Diaion HP-20 CC eluted with MeOH/H2O (2 : 3) and finally Me2CO/EtOH (1 : 1) to obtain sugar fractions (3.3 mg from 1; 0.79 mg from 2; 1.4 mg from 3; 2.9 mg from 4; 0.77 mg from 5; 0.38 mg from 6; 4.5 mg from 7; 2.8 mg from 8; 2.3 mg from 9; 0.92 mg from 10; 0.8 mg from 11; 2.3 mg from 12; 2.0 mg from 13; 2.5 mg from 14; 2.6 mg from 15; 2.1 mg from 16; 0.94 mg from 17; 3.8 mg from 18; 1.9 mg from 19; and 1.0 mg from 20). The sugar fractions were analyzed by HPLC under the following conditions: column, Capcell Pak NH2 UG80 (4.6 mm i.d. × 250 mm, 5 µm, Shiseido, Tokyo, Japan); solvent, MeCN/H2O (17 : 3); detection, refractive index and optical rotation; flow rate, 1.0 mL/min. D-Apiose in 3, 4, 8–12, and 14–19, L-arabinose in 1–20, D-fucose in 4, 7, 10, 13, 15, and 16, D-galactose in 11–15, D-glucose in 5 and 13–15, D-quinovose in 2, L-rhamnose in 1–20, and D-xylose in 1–20 were identified by comparing their retention times (tR) and optical rotations with those of authentic samples: D-apiose (6.66, positive optical rotation), L-rhamnose (7.64, negative optical rotation), D-fucose (8.42, positive optical rotation), D-quinovose (8.78, positive optical rotation), L-arabinose (9.57, positive optical rotation), D-xylose (9.92, positive optical rotation), D-galactose (15.63, positive optical rotation), and D-glucose (16.14, positive optical rotation).
Enzymatic Hydrolysis of 21Compound 21 (15.0 mg) was treated with naringinase (45.0 mg, EC 232-962-4, Sigma) in AcOH/AcOK buffer (pH 4.3, 5.0 mL) at 28°C for 84 h. The reaction mixture was purified by silica gel CC using CHCl3/MeOH/H2O (90 : 10 : 1) to yield 21a (12.2 mg) and a sugar fraction (1.3 mg). HPLC analysis of the sugar fraction under the same conditions as those of 1 showed the presence of D-glucose (16.14, positive optical rotation).
Cytotoxic Activity AssayHL-60 and A549 cells were kept in RPMI-1640 medium and MEM, respectively. These cell media contained 100 µg/mL streptomycin sulfate, penicillin G sodium salt, and heat-inactivated 10% FBS supplemented with L-glutamine. Cytotoxic activity of the isolated compounds against HL-60 and A549 cells was evaluated by a modified MTT assay method as previously reported.24)
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.