Notes
Syntheses of C2′-Fluorinated Analogs of Solamin
2020 年 68 巻 7 号 p. 675-678
詳細
2020 年 68 巻 7 号 p. 675-678
The details of the total syntheses of C2′-fluorinated analogs of solamin, an antitumor annonaceous acetogenin, are described. Fluorine was enantioselectively introduced at the C2′-position by organocatalytic α-fluorination of the aldehyde according to a previously reported method. C2′-fluorinated solamin and its C2′-diastereomer were synthesized by the Sonogashira coupling of a tetrahydrofuran fragment and fluorine-containing γ-lactone fragments.
The fluorination of organic molecules is a powerful tool for the development of novel drugs and agrichemicals. In medicinal chemistry, the fluorine atom is of particular interest due to its similar size to the hydrogen atom, its high electron-withdrawing ability, the greater stability of the C–F bond compared to the C–H bond, and its influence on the lipophilicity of a molecule.1) These properties provide various benefits, such as improved metabolic stability, altered physicochemical properties, and increased binding affinity.
Annonaceous acetogenins are polyketides isolated from the Annonaceae plant that grows in tropical and sub-tropical regions. More than 500 acetogenins have been isolated to date, with the majority possessing long hydrocarbon chains bearing one to three adjacent or non-adjacent 2,5-disubstituted tetrahydrofuran (THF) moieties at their molecular centers, in addition to an α,β-unsaturated-γ-lactone ring moiety at the end of each molecule.2–6) Acetogenins are known as potent inhibitors of mitochondrial reduced nicotinamide-adenine dinucleotide (NADH) ubiquinone oxidoreductase (complex I),7–9) and chemists have been attracted to acetogenins because of their unique chemical structures and broad biological activities, which include immunosuppressive, antimalarial, and antifeedant properties, in addition to cytotoxicity against human cancer cells. Although many total syntheses of natural acetogenins10) and their analogs11–18) have been reported, the preparation of their fluorinated analogs have remained unexamined until our report into the synthesis of C2′-fluorinated solamin.19–21)
Solamin is a mono-THF acetogenin with a simple structure, but contains all of the components that characterize acetogenins22–24) (Fig. 1). Murisolin, a C2′-hydroxy solamin, was isolated from the same plant as solamin, namely Annona muricata. Although the two structures are identical, except for the hydroxy group at the C2′ position in the latter, their growth inhibitory activities against human cancer cell lines differ significantly. More specifically, murisolin inhibits the growth of DMS114, a human lung cancer cell line, 400-times more strongly than solamin. We were therefore interested in the biological activity of C2′-fluorinated solamin 1, since the fluorine atom is known to mimic the hydrogen atom and can also potentially function in a similar manner to the hydroxy group due to its high electron density. In fact, the introduction of a fluorine atom at the C2′ position was previously reported to increase growth inhibitory activity against human cancer cell lines compared to solamin, albeit to a lesser extent than the activity displayed by murisolin.19)

In this context, functionalizing the C2′ positions of acetogenins is an attractive research topic; however, to the best of our knowledge, only a single report has been published into C2′-chlorinated analogs, which were synthesized by the Appel reactions of natural C2′-hydroxy acetogenins.25) Recently, some efficient late-stage deoxy-fluorinations of secondary alcohols have also been reported26,27); however, it may be difficult to apply this method to the syntheses of C2′-fluorinated acetogenins, since natural acetogenins contain highly reactive chiral α,β-unsaturated-γ-lactone moieties that are easily epimerized.28) With this in mind, we herein report the details of the syntheses of C2′-fluorinated solamin 1 and its C2′-diastereomer.
Our approach to C2′-fluorinated analog 1 is outlined retrosynthetically in Chart 1. More specifically, analog 1 can be synthesized by the Sonogashira coupling29) of alkyne 2, prepared by our stereodivergent synthetic route,30–37) and vinyl iodide 3. The γ-lactone fragment 3 can be obtained by the α-alkylation of the known α-sulfenyl-γ-lactone 538) with iodide 4. We planned to construct the chiral center of 4 by the enantioselective α-fluorination of known aldehyde 6.39)

TBS = tert-butyl(dimethyl)silyl.
Table 1 summarizes the results obtained during the optimization of the enantioselective α-fluorination of known aldehyde 6. The enantioselective organocatalytic α-fluorination of aldehydes has previously been developed by Beeson and MacMillan40) and was used in this study. The resulting α-fluoro aldehyde was isolated as the β-fluoro alcohol 7 after reduction due to the instability of the α-fluoro aldehyde. According to the reported procedure, aldehyde 6 (E/Z = 85 : 15) was treated with 5.0 equiv. of N-fluorobenzenesulfonimide (NFSI) in the presence of 20 mol% (R)-5-benzyl-2,2,3-trimethylimidazolidin-4-one dichloroacetic acid salt [(R)-8] at −10°C, followed by reduction with NaBH4 to give 7 in 53% yield over two steps (entry 1). Both higher and lower reaction temperatures were found to produce lower yields (entries 2–3). Although the reaction with 1.0 equiv. of NFSI afforded an inseparable mixture of 7 and 5-iodopent-4-en-1-ol from unreacted 6 (entry 5), a similar yield to that of entry 1 and high enantioselectivity were achieved when 2.5 equiv. of NFSI was employed (entry 4). Using the optimized conditions, a larger scale reaction gave enantiomerically pure 7 in 50% yield (entry 6).41)
 | ||||
|---|---|---|---|---|
| Entrya) | NFSI (eq.) | Temp. (°C) | Time (h) | Yield (% over two steps)b) |
| 1 | 5.0 | −10 | 24 | 53 |
| 2 | 5.0 | −20 | 47 | 39 |
| 3 | 5.0 | 0 | 10 | 42 |
| 4 | 2.5 | −10 | 33 | 52 (>98%ee)c) |
| 5 | 1.0 | −10 | 48 | 51d) |
| 6e) | 2.5 | −10 | 24 | 50 (>98%ee)c) |
a) Unless otherwise stated, the reaction was performed using aldehyde 6 (0.2–0.3 mmol). b) Isolated yield. c) The enantiomeric excess (ee) was determined by 1H-NMR analysis of the corresponding Mosher ester of 7.42,43)d) Yield was determined by 1H-NMR analysis. e) The reaction was carried out using 5.6 mmol of aldehyde 6.
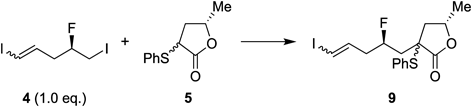
With alcohol 7 in hand, the Appel reaction of this compound was carried out in N,N-dimethylformamide (DMF) at 100°C to give the corresponding iodide 4 in high yield. The reaction conditions for the α-alkylation of α-sulfenyl-γ-lactone 5 with iodide 4 were optimized, the results of which are presented in Table 2. More specifically, the coupling reaction of 1.0 equiv. of 4 with 2.0 equiv. of lactone 5 was carried out with lithium diisopropylamide (LDA) in the presence of hexamethylphosphoramide (HMPA), which are the standard reaction conditions reported for a similar substrate devoid of the fluorine atom β to the leaving group,44) to give the desired product 9 in moderate yield (entry 1). No product was obtained in the absence of HMPA or when HMPA was used with potassium hexamethyldisilazide (KHMDS) as the base (entries 2–3), while the use of t-BuOK in DMF at room temperature (r.t.)45) gave the same result as that of entry 1 (entry 4). We further investigated the reagent handling conditions, which resulted in good product yields when 4.0 equiv. of 5 was mixed with t-BuOK in DMF or dimethyl sulfoxide (DMSO) at r.t. (entries 5–6). Finally, oxidation of sulfide 9 with m-chloroperoxybenzoic acid (m-CPBA) followed by thermal elimination of the resulting sulfoxide in toluene afforded a good yield of the E-form of α,β-unsaturated-γ-lactone 10 bearing a fluorine atom at the desired position.46)
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Conditions | Yield (%) | |||||
| 5 (eq.) | Base | Solv. | HMPA (eq.) | Temp. | Time (h) | ||
| 1 | 2.0 | LDA | THF | 2.0 | −78 to 0°C | 5 | 47 |
| 2 | 2.0 | LDA | THF | − | −78 to 0°C | 5 | Not detected |
| 3 | 2.0 | KHMDS | THF | 2.0 | 0°C to r.t. | 5 | Complex mixture |
| 4 | 2.0 | t-BuOK | DMF | − | r.t. | 20 | 43 |
| 5 | 4.0 | t-BuOK | DMF | − | r.t. | 20 | 72 |
| 6 | 4.0 | t-BuOK | DMSO | − | r.t. | 20 | 74 |

Reagents and conditions: (a) I2, PPh3, DMF, 100°C, 90%; (b) m-CPBA, CH2Cl2, 0°C; (c) toluene, reflux, 70% over two steps.
The C2′-epimer 12 was also prepared in good yield using the antipodal MacMillan’s catalyst according to the procedure employed for the synthesis of 10 (Chart 3).

Reagents and conditions: (a) NFSI, (S)-8, THF/i-PrOH, −10°C; (b) NaBH4, CH2Cl2/EtOH, r.t., 66% (>98%ee) over two steps; (c) I2, PPh3, DMF, 100°C, 88%; (d) 5, t-BuOK, DMF, r.t., 79%; (e) m-CPBA, CH2Cl2, 0°C; (f) toluene, reflux, 70% over two steps.
1H-NMR spectroscopy was used to determine the diastereomeric purities at the fluorine-bearing carbon atoms of 10 and 12 (Table 3). Fortunately, both compounds exhibited differences in the chemical shifts of the protons at the C2′ and C1″ positions in C6D6, although the same pattern of chemical shifts was observed in CDCl3. The results revealed that diastereomerically pure 10 and 12 had been obtained, although it is unclear why there are differences in the chemical shifts in C6D6 since flexibility exists around the C2′ positions in both compounds.
 | ||||
|---|---|---|---|---|
| Position | δ (ppm) | |||
| in C6D6 | in CDCl3 | |||
| 10 | 12 | 10 | 12 | |
| 4 | 6.20 | 6.20 | 7.24 | 7.24 |
| 5 | 4.18 | 4.17 | 5.07 | 5.06 |
| 1′ | 2.02–2.18 | 2.04–2.17 | 2.52–2.64 | 2.51–2.64 |
| 2′ | 4.26 | 4.29 | 4.79 | 4.78 |
| 3′ | 1.64–1.81 | 1.65–1.81 | 2.40–2.47 | 2.40–2.47 |
| 4′ | 6.25 | 6.25 | 6.56 | 6.56 |
| 5′ | 5.71 | 5.71 | 6.24 | 6.24 |
| 1″ | 0.78 | 0.75 | 1.44 | 1.44 |
With the γ-lactone fragments in hand, the Sonogashira coupling of the THF-ring fragment 2 with 10 gave enediyne 13 in good yield (Chart 4). Finally, the selective reduction of the enediyne moiety of 13 with a diimide,47) followed by deprotection of the TBS group under acidic conditions, gave the desired C2′-fluorinated solamin 1. The diastereomer at the C2′ position (17) was obtained in a similar manner by replacing 10 with 12 in the described Sonogashira-coupling reaction.

Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI, Et3N, r.t., 84% from 10, 77% from 12; (b) p-toluenesulfonyl hydrazide, NaOAc, 1,2-dimethoxyethane/H2O, reflux, 56% from 13, 69% from 15; (c) 48% HF aq., CH3CN/THF, r.t., 88% from 14, 83% from 16.
Convergent syntheses of C2′-fluorinated analogs of solamin were accomplished, in which a fluorine atom was successfully introduced into the C2′ position by the enantioselective α-fluorination of an aldehyde using the procedure developed by MacMillan. Sonogashira coupling of the THF-ring fragment and the γ-lactone fragment afforded the desired C2′-fluorinated analog. We are currently undertaking further research to establish structure–activity relationships of such attractive fluorinated analogs of the annonaceous acetogenins.
Melting points are uncorrected. Optical rotations were measured using a JASCO DIP-360 digital polarimeter or a JASCO P-1020 digital polarimeter. 1H-NMR spectra were recorded in the specified solvent with a JEOL JNM-GX-500 spectrometer (500 MHz), a JEOL JNM-AL300 spectrometer (300 MHz), or a JEOL JNM-EX-270 spectrometer (270 MHz). 13C-NMR spectra were recorded in the specified solvent with a JEOL JNM-AL300 spectrometer (75 MHz). Chemical shifts are reported in ppm relative to the internal solvent signal [CDCl3: 7.26 ppm (1H-NMR), 77.0 ppm (13C-NMR)] or tetramethylsilane [0 ppm] as the internal standard. The following abbreviations are used: broad singlet = br s, singlet = s, doublet = d, triplet = t, quartet = q, quintet = qn, sextet = sext, septet = sep, and multiplet = m. IR absorption spectra (FT = diffuse reflectance spectroscopy) were recorded with KBr powder using a Horiba FT-210 IR spectrophotometer, or as neat films on NaCl plates using a Shimadzu FTIR-8400S instrument, and only noteworthy absorptions (in cm−1) are listed. Mass spectra were recorded on JEOL JMS-600H and JEOL JMS-700 mass spectrometers. Column chromatography was carried out using Kanto Chemical Silica Gel 60 N (spherical, neutral, 63–210 µm), and flash column chromatography was carried out using Merck Silica Gel 60 (40–63 µm). All air- or moisture-sensitive reactions were carried out in flame-dried glassware under an atmosphere of Ar or N2. All solvents were dried and distilled according to standard procedures, if necessary. All organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure with a rotary evaporator.
This work was supported in part by JSPS KAKENHI [Grant Numbers 25460159, 16K08330] (N.K.), the Ministry of Education, Culture, Sports, Science and Technology (MEXT)-Supported Program for the Strategic Research Foundation at Private Universities, 2015–2019 [Grant Number S1511024L] (N.K.), and a Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research (N.K.).
The authors declare no conflicts of interest.
The online version of this article contains supplementary materials. See supplementary materials for details regarding compound preparation and characterization.