Results and Discussion
Inhibitor ScreeningThe targeted Golgi β-galactosidase activity was previously observed in three human cell lines, HeLa cells, HT-1080 cells, and SK-N-SH cells, using QMC platform-based fluorogenic substrates.12) To construct the cell-based inhibitor HTS system in a 96-well format to identify inhibitors, we used HeLa cells and our developed fluorogenic substrate 1 incorporating 2-methyl TokyoGreen (2MeTG) as the fluorophore. Substrate 1 has an acetyl modification that provides cell-membrane permeability.11–14) The structure of the substrate used in this study is shown in Fig. 1. We constructed the 96-well format inhibitor HTS system based on fluorescence intensity (FI) changes due to substrate 1 hydrolysis by the Golgi β-galactosidase in cultured HeLa cells, according to our previous report,13) with modifications made to optimize the target enzyme activity. The quality and suitability of the constructed HTS system were evaluated based on the Z′ factor, the coefficient of variation (CV), and the signal-to-background ratio (S/B).13,19) The Z′ factor for the system was calculated to be 0.72 using the FI from substrate 1 based on evaluation of the signal and background control levels. In addition, the calculated inter-well variability CV was < 8.2%, and the obtained S/B was 11.9. Together, these statistical parameters confirm the high quality and suitability of the system. The HTS system is useful for primary inhibitor screening against a compound library that especially requires high screening speed and efficiency.
The constructed cell-based HTS system was used to primarily screen two compound libraries for the Golgi β-galactosidase inhibitors, namely a natural compound library (NPDepo library, RIKEN) and an herbal medicine library (Institute of Natural Medicine, University of Toyama). In the primary HTS screen, we used three times the value of the standard deviation (S.D.) + average (Ave) of the inhibition rate for each inhibitor as the standard hit selection criteria and a cutoff parameter. Any compound that exhibited a more significant percentage inhibition than the cutoff parameter was declared nominally active. The first hits included 50 compounds that decreased <3SD + Ave for the negative control. In the next inhibitor screen, we eliminated compounds yielding false-positive hits from the 50 primary hit compounds. The Z′ value of the inhibitor HTS was sufficiently high to exclude false-positive hit compounds; however, eliminating non-specific and/or covalent enzyme inhibitors is challenging. To remove the false-positive hit compounds from the 50 compounds, they were tested in dose–response experiments in a secondary screen using the same HTS system. Of these, 22 compounds showed a dose–response inhibition. The details of the primary and secondary screening conditions are described in Experimental.
The HTS system did not enable further selection of secondary hit compounds as the inhibitory activity would include false positives that was mainly based on cytotoxicity. Therefore, a third screening was performed in vitro against the hits from the second screen using commercial rpGLB1 (AAs 24–677 with a C-terminal 6-His tag) and substrate 2 (Fig. 1). In the third screen, the inhibitory activity was evaluated solely as the affinity of the inhibitor for rpGLB1. To this end, we first confirmed the β-galactosidase activity of rpGLB1 using substrate 2 at two pH values, pH 3.5 in lysosomes and pH 6.5 in the Golgi apparatus. The enzyme activities were evaluated by observing the FI increase due to 2MeTG release at 37°C for 60 min at 30 s intervals. The results revealed the relative hydrolysis rates, which were normalized to the amount of FI released from substrate 2 at pH 6.5 (set as 100%). Our results indicated that rpGLB1 exhibited 13.1% hydrolyzing activity at pH 3.5. Thus, rpGLB1 showed approximately eight-fold higher hydrolyzing activity at pH 6.5 than at pH 3.5. Presumably, the higher activity at pH 6.5 reflected the increased quantum yield of the fluorophore at an almost neutral pH, as well as differences in the conformation of rpGLB1 at either pH. Therefore, the third screening against 22 hits from the second screen was performed using rpGLB1, which had sufficient enzymatic activity at the pH of the Golgi apparatus. The inhibitory activity was evaluated by observing the decrease in FI of 2MeTG released from substrate 2 by rpGLB1 in the presence of each compound at 20 µM at a pH of 6.5. Next, the two top hits from the NPDepo library (ARM00 and 2–3F09) and the herbal medicine library (Hirsutine and Shikonin) were selected for further study. The structures of these inhibitors are summarized in Fig. 2. Furthermore, the all compounds were tested in dose–response experiments at concentrations of 0.5, 12.5, and 20 µM, using the same in vitro system. All compounds except for Shikonin, exhibited a dose-dependent FI inhibition. The conditions used in the third screen are described in Experimental.
Because effective inhibitors act as affinity-based chemical probes at the cellular level, it was necessary to probe whether any of the three inhibitors might interact with off-target proteins in human cells. Therefore, the cytotoxicity of the compounds was evaluated using the WST-8 method as a final screening method according to our previous report, with slight modifications.20) The three hit compounds, ARM00, 2–3F09, and Hirsutine, were assessed for cytotoxicity at a final concentration of 100 µM for 24 h, revealing cell survival rates of 67.8, 11.1, and 24.3%, respectively. Based on these results, we selected ARM00 as the final hit compound from among the candidate Golgi β-galactosidase inhibitors. ARM00 has been reported as one of the components of a patented sweetener,21) and no reports are available describing its synthesis method and biological activity.
Design and Synthesis of the InhibitorStructurally, ARM00 has a slightly modified isoflavone scaffold. We decided to preliminarily investigate its SAR to develop more potent inhibitors. To determine the optimal chlorine substitution position of ARM00, we evaluated the rpGLB1 inhibitory activity of three commercially available positional isomers, o-isomer (ARM00), m-isomer (ARM01), and p-isomer (ARM02), at 75 µM. Moreover, the rpGLB1 inhibitory activities of three commercial compounds, ARM03–ARM05 (in which a trifluoromethyl group was introduced at the 2-position of the isoflavone skeleton of ARM00–ARM02 to enhance their interactions for pGLB1), were evaluated at 75 µM. Our results showed that the highest inhibitory activity was observed when the chlorine was substituted at the o-position. However, introducing a trifluoromethyl group into ARM00–ARM02 decreased the inhibitory activity. Briefly, none of the evaluated derivatives had greater inhibitory activity than ARM00. The structures of ARM00–ARM05 and their inhibitory activities are summarized in Fig. 3.
To enhance the inhibitory activity of ARM00, the binding mode of ARM00 to the active site of the enzyme need to be identified. However, the preliminary SAR study yielded little information. The molecular basis of the observed inhibition of pGLB1 by ARM00 was investigated by performing molecular docking simulations, which can effectively predict predominant the binding modes of a ligand located within an active site. We performed molecular docking simulations using the crystallographic structure of pGLB1 (AAs 24–677, PDB ID: 3THD,22)) and ARM00 as a ligand. In the crystallographic structure, DGJ is bound to subsite −1 at the active site with a proper degree of packing (Fig. 4A). In the docking model, ARM00 was docked in the active site, with the 2-chlorobenzyl moiety being fully packed at subsite −1 (Fig. 4B).
Therefore, we designed different derivatives with substituents of appropriate sizes for subsite −1: 3-methylbutyl (ARM06), 3-methyl-2-butenyl (ARM07), and 3-methyl-2-butynyl (ARM08). The inhibitory activities of these derivatives were expected to exceed those of ARM00. We also designed ARM09 with a methyl group as a too-small substituent and ARM10 with a 4-phenylbenzyl group as a too-large substituent, and their inhibitory activities were predicted to be lower than those of ARM00. In addition, ARM11 was designed to alleviate overcrowding at subsite −1 by removing the chlorine atom of ARM00, which could potentially increase the inhibitory activity. The initial SAR studies of these six designed derivatives were expected to reveal the binding mode of the ARM00 derivatives to the active site. However, synthesis methods for ARM00 and ARM06–ARM11 have not been reported, we synthesized these derivatives, according to previous reports of similar compounds.23,24,25) The structure and synthetic scheme of ARM00 and ARM06–ARM11 are summarized in Fig. 5. The 4-hydroxyl group of 2,4-dihydroxyacetophenone, as the starting material, was selectively protected by a tetrahydropyranyl (THP) group to obtain compound 1. Compound 1 was condensed with N,N-dimethylformamide dimethylacetal to obtain compound 2. The crude compound 2 was directly treated with I2 to furnish compound 3, which was cross-coupled with 2-methoxyphenylboronic acid by solution-phase Suzuki–Miyaura reaction to obtain compound 4. Cleavage of the THP group of 4 under acidic conditions yielded the common intermediate 5. Finally, alkylation of the phenol of 5 with corresponding alkyl halides gave ARM00 and ARM06–ARM11. All synthesized derivatives were characterized by 1H-NMR, 13C-NMR, MS analysis, and elemental analysis.
Biological StudyFirst, we evaluated the rpGLB1 inhibitory activities of ARM00, ARM06–ARM11, and DGJ at 25 µM. The inhibition rates of ARM00 as an original compound and DGJ as a reference compound were 50.2 and 65.3%, respectively. The inhibition rates of ARM06–ARM08 in successful binding to the active site were 51.7, 71.3, and 30.4%, respectively. ARM09 and ARM10 showed the low inhibition rates of 6.1 and 20.0%, respectively, in preventing binding to the active site. ARM11 showed lower inhibitory activity than ARM00 (41.1 vs. 50.2%), despite reducing any potential steric hindrance at subsite −1. This reduction in inhibitory activity indicates that subsite −1 contains a recognition site for the substituent at the o-position. Therefore, appropriate structural modification of the o-position of the inhibitor may lead to an increase in the inhibitory activity. ARM07 was the most potent rpGLB1 inhibitor, including the original and reference compounds.
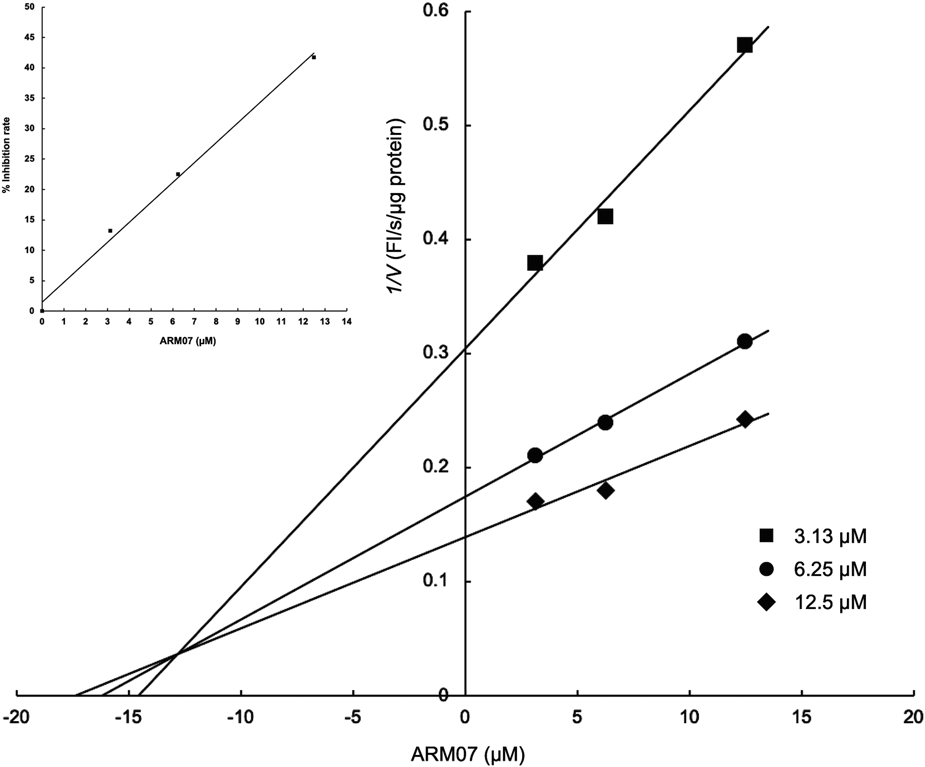
Based on these results, the ARM00 derivatives are predicted to act as reversible inhibitors of rpGLB1 through a competitive inhibition mechanism, and the 2-chlorobenzyl moiety of ARM00 and the corresponding moieties of ARM06–ARM08 and ARM11 are predicted to bind to subsite −1 at the active site. These possibilities can be tested by determining the enzyme-inhibition type and kinetic parameters of the ARM00 derivatives. Therefore, the most potent inhibitor ARM07 was selected to determine the mechanism of this inhibition type and the kinetic parameters, IC50 value and Ki value. The inhibition rates of ARM07 for rpGLB1 plotted in a dose–response linear curves led to an IC50 value of 14.8 µM (Fig. 6, inset). A Dixon plot was generated to derive the Ki value (Fig. 6). The plot had a common intersection at the orthant II, indicating pure competitive inhibition of rpGLB1 by ARM07, i.e., that ARM07 bound the enzyme at the active site. The Ki value was calculated directly from the plot and was confirmed by plotting the 1/V (FI/s/µg protein) against different ARM07 concentrations. The obtained Ki value was 13.3 µM, demonstrating that ARM07 has high affinity for rpGLB1.
Next, we evaluated the Golgi β-galactosidase inhibitory activities of ARM07 and DGJ in HeLa cells in a 6-well plate. The inhibitory activities of both inhibitors were assessed by measuring changes in fluorescence signals due to enzymatic hydrolysis of substrate 1. Substrate 1 was added to the HeLa cells a final concentration of 10 µM, ARM07 was added to a final concentration of 50 µM, or DGJ was added to a final concentration of 300 µM, followed by incubation for 2 h. Next, the fluorescence signals from each well were recorded using a fluorescence microscope, and then the FI and fluorescence area (FA) of the obtained images were analyzed using an image-analysis software. The inhibition rates were calculated according to previous reports, with slight modifications.13,20) ARM07 and DGJ inhibited the FI by 74.0 and 60.9%, and the FA by 31.5 and 25.2%, respectively, indicating that ARM07 exhibited approximately more than twice inhibitory activity at a one-sixth concentration of DGJ and almost chemically knocked down the Golgi β-galactosidase activity in living HeLa cells. Moreover, ARM07 showed no cytotoxicity under these conditions. The low inhibitory activity of DGJ could be explained by its low hydrophobicity necessitating the use of a high working concentration based on its low membrane permeability. Taken together, these findings suggest that ARM07 has a high affinity for the Golgi β-galactosidase and exhibits no off-target proteins that cause cytotoxicity in HeLa cells. These considerations are of paramount importance in the development of affinity-based chemical probes that could identify the protein responsible for the Golgi β-galactosidase activity.
Experimental
ChemistryNew compounds were characterized by 1H-NMR, 13C-NMR, 1H–1H correlation spectroscopy, heteronuclear multiple quantum coherence spectrometry, MS, and elemental analysis. The NMR spectra were recorded with a JEOL ECA500 spectrometer (JEOL, Japan; 500 MHz for 1H and 125 MHz for 13C). Chemical shifts were expressed in ppm as downfield shifts from Me4Si. Low-resolution mass spectra were obtained with an LCMS-2020 (Shimadzu, Japan) mass spectrometer, and DUIS-2020 Dual Ion Source (DUIS) probe, which was coupled to a LC-2030C 3D (Shimadzu, Japan), was used for simultaneous electrospray ionization and atmospheric pressure chemical ionization measurements. Column chromatography was performed using Silica Gel 60N (spherical neutral particle size: 100–210 µm, Kanto Chemical, Japan). The progress of all reactions was monitored by TLC on Silica Gel 60 F254, 0.25 mm (Merck KGaA, Darmstadt, Germany). Melting points were determined with a Yamato Model MP-21 capillary apparatus and are uncorrected. ARM01-ARM05 were purchased from NAMIKI SHOJI Co., Ltd. (Japan). Compounds 1–5 were synthesized as described previously, with slight modifications.23) The NMR and DUIS-MS data for compounds 1,23)3,23)4,24) and 5,25) were consistent with previously reported data.
General Synthetic Procedure for ARM00 and ARM06–ARM11Common intermediate 5 (100 mg, 1 equivalent (equiv.)) was dissolved in 1 mL of acetone. Corresponding alkyl halide (2 equiv.) and potassium carbonate (3 equiv.) were added, the reaction mixture was stirred at ambient temperature for 1 to 4 h, the precipitate was removed by Kiriyama filtration, and the solvent was concentrated. The obtained residue was recrystallized from hot ethanol or purified by column chromatography on silica gel (hexane : ethyl acetate = 2 : 1).
7-(2-Chlorobenzyloxy)-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM00)White crystalline solid (0.10 g, 68%); melting point 130–131°C; 1H-NMR (CDCl3) δ: 3.80 (3H, s, 2′-OCH3), 5.28 (2H, s, benzyl position), 6.95 (1H, d, J = 2.0 Hz, H-8), 6.99 (1H, d, J = 8.5 Hz, H-6′), 7.02 (1H, dd, J = 1.0 Hz, J = 7.5 Hz, H-4′), 7.09 (1H, dd, J = 2.0 Hz, J = 8.8 Hz, H-6), 7.31–7.34 (3H, m, H-5′, H-3″, and H-6″), 7.37 (1H, m, H-3′), 7.44 (1H, m, H-4″), 7.55 (1H, m, H-5″), 7.91 (1H, s, H-2), 8.23 (1H, d, J = 8.8 Hz, H-5); 13C-NMR (CDCl3) δ: 55.76 (2′-OCH3), 67.59 (benzyl position), 101.22 (C-8), 111.22 (C-6′), 114.78 (C-6), 118.88 (C-10), 120.56 (C-4′), 120.90 (C-1′), 122.67 (C-3), 127.13 (C-6″), 128.03 (C-5), 128.82 (C-5″), 129.45 (C-4″), 129.60 (C-3″), 129.77 (C-3′), 131.75 (C-5′), 132.76 (C1″ or C2″), 133.53 (C1″ or C2″), 153.83 (C-2), 157.54 (C-2′), 157.91 (C-9), 162.58 (C-7), 175.45 (–C=O); DUIS-MS (positive mode): m/z = 393 [M + H]+, (negative mode): m/z = 391 [M–H]−. Anal. Calcd for C23H17ClO4: C, 70.32; H, 4.36; N, 0.00; Cl, 9.02; O, 16.29. Found: C, 70.33; H, 4.36; N, 0.00.
7-(3-Methylbutyloxy)-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM06)White solid (11 mg, 18%); 1H-NMR (CDCl3) δ: 0.99 (6H, d, J = 7.0 Hz, –CH(CH3)2), 1.74 (2H, td, J = 6.5 Hz, J = 7.0 Hz, –OCH2CH2CH(CH3)2), 1.87 (1H, qd, J = 7.0 Hz, –OCH2CH2CH(CH3)2), 3.80 (3H, s, 2′-OCH3), 4.09 (2H, t, J = 6.5 Hz, –OCH2CH2CH(CH3)2), 6.85 (1H, d, J = 2.5 Hz, H-8), 6.97 (1H, dd, J = 2.5 Hz, J = 8.5 Hz, H-6), 6.98 (1H, d, J = 7.5 Hz, H-6′), 7.03 (1H, dd, J = 7.5 Hz, H-4′), 7.31 (1H, dd, J = 1.5 Hz, J = 7.5 Hz, H-5′), 7.36 (1H, ddd, J = 2.0 Hz, J = 7.5 Hz, H-3′), 7.90 (1H, s, H-2), 8.16 (1H, d, J = 8.5 Hz, H-5); 13C-NMR (CDCl3) δ: 22.55 (–CH(CH3)2), 25.04 (–OCH2CH2CH(CH3)2), 37.66 (–OCH2CH2CH(CH3)2), 55.77 (2′-OCH3), 67.08 (–OCH2CH2CH(CH3)2), 100.62 (C-8), 111.23 (C-6′), 114.72 (C-6), 118.31 (C-10), 120.55 (C-4′), 121.03 (C-1′), 122.57 (C-3), 127.75 (C-5), 129.71 (C-3′), 131.78 (C-5′), 153.74 (C-2), 157.55 (C-2′), 158.03 (C-9), 163.43 (C-7), 175.52 (–C=O); DUIS-MS (positive mode): m/z = 339 [M + H]+; Anal. Calcd for C21H22O4: C, 74.54; H, 6.55; N, 0.00; O, 18.91. Found: C, 74.54; H, 6.41; N, 0.00.
7-(3-Methyl-2-butenyloxy)-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM07)White crystalline solid (76 mg, 61%); melting point 133–134°C; 1H-NMR (CDCl3) δ: 1.79 (3H, s, =C(CH3)2), 1.83 (3H, s, –C(CH3)2), 3.80 (3H, s, 2′-OCH3), 4.62 (2H, d, J = 6.5 Hz, –OCH2CH=C(CH3)2), 5.50–5.53 (1H, m, –OCH2CH=C(CH3)2), 6.86 (1H, d, J = 2.5 Hz, H-8), 6.98 (2H, dd ×2, J = 2.5 Hz, J = 8.5 Hz, H-6′ and H-6), 7.02 (1H, ddd, J = 1.0 Hz, J = 7.5 Hz, H-4′), 7.32 (1H, dd, J = 1.5 Hz, J = 7.5 Hz, H-5′), 7.36 (1H, ddd, J = 1.5 Hz, J = 7.5 Hz, H-3′), 7.90 (1H, s, H-2), 8.19 (1H, d, J = 8.5 Hz, H-5); 13C-NMR (CDCl3) δ: 18.31 (=C(CH3)2), 25.86 (–C(CH3)2), 55.77 (2′-OCH3), 65.42 (–OCH2CH=C(CH3)2), 100.92 (C-8), 111.22 (C-6′), 114.83 (C-6), 118.38 (C-10), 118.62 (–OCH2CH=C(CH3)2), 120.55 (C-4′), 121.00 (C-1′), 122.58 (C-3), 127.77 (C-5), 129.72 (C-3′), 131.78 (C-5′), 139.34 (=C(CH3)2), 153.75 (C-2), 157.55 (C-2′), 157.96 (C-9), 163.12 (C-7), 175.52 (–C=O); DUIS-MS (positive mode): m/z = 337 [M + H]+, 359 [M + Na]+, (negative mode): m/z = 335 [M–H]−; Anal. Calcd for C21H20O4: C, 74.98; H, 5.99; N, 0.00; O, 19.02. Found: C, 74.84; H, 5.73; N, 0.00.
7-(3-Methyl-2-butynyloxy)-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM08)White crystalline solid (53 mg, 44%); melting point 116–117°C; 1H-NMR (CDCl3) δ: 1.89 (3H, t, J = 2.0 Hz, –OCH2C≡C–CH3), 3.80 (3H, s, 2′-OCH3), 4.76 (2H, q, J = 2.0 Hz, –OCH2C≡CCH3), 6.97–7.01 (2H, m, H-8 and H-6′), 7.02 (1H, ddd, J = 1.0 Hz, J = 7.5 Hz, H-4′), 7.03 (1H, dd, J = 2.8 Hz, J = 9.0 Hz, H-6), 7.32 (1H, dd, J = 1.5 Hz, J = 7.5 Hz, H-5′), 7.37 (1H, ddd, J = 1.5 Hz, J = 7.5 Hz, H-3′), 7.92 (1H, s, H-2), 8.21 (1H, d, J = 9.0 Hz, H-5); 13C-NMR (CDCl3) δ: 3.73 (–C≡CCH3), 55.76 (2′-OCH3), 56.92 (–OCH2C≡CCH3), 72.95 (–C≡C–), 84.89 (–C≡C–), 101.51 (C-8), 111.21 (C-6′), 114.77 (C-6), 118.84 (C-10), 120.56 (C-4′), 120.92 (C-1′), 122.66 (C-3), 127.85 (C-5), 129.77 (C-3′), 131.75 (C-5′), 153.83 (C-2), 157.55 (C-2′), 157.80 (C-9), 161.93 (C-7), 175.51 (–C=O); DUIS-MS (positive mode): m/z = 321 [M + H]+, 343 [M + Na]+; Anal. Calcd for C20H16O4: C, 74.99; H, 5.03; N, 0.00; O, 19.98. Found: C, 74.99; H, 5.20; N, 0.00.
7-Methoxy-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM09)White crystalline solid (91 mg, 86%); melting point 109–110°C; 1H-NMR (CDCl3) δ: 3.80 (3H, s, 2′-OCH3), 3.92 (3H, s, 7-OCH3), 6.86 (1H, d, J = 2.0 Hz, H-8), 6.99 (2H, dd x 2, J = 2.0 Hz, J = 8.5 Hz, H-6′ and H-6), 7.02 (1H, dd, J = 7.5 Hz, H-4′), 7.32 (1H, dd, J = 1.5 Hz, J = 7.5 Hz, H-5′), 7.37 (1H, ddd, 1H, J = 1.5 Hz, J = 7.5 Hz, H-3′), 7.91 (1H, s, H-2), 8.20 (1H, d, J = 8.5 Hz, H-5); 13C-NMR (CDCl3) δ: 55.77 (2′-OCH3), 55.82 (7-OCH3), 100.16 (C-8), 111.23 (C-6′), 114.35 (C-6), 118.50 (C-10), 120.56 (C-4′), 120.97 (C-1′), 122.63 (C-3), 127.84 (C-5), 129.74 (C-3′), 131.77 (C-5′), 153.77 (C-2), 157.55 (C-2′), 158.02 (C-9), 163.87 (C-7), 175.50 (–C=O); DUIS-MS (positive mode): m/z = 283 [M + H]+, 305 [M + Na]+; Anal. Calcd for C17H14O4: C, 72.33; H, 5.00; N, 0.00; O, 22.67. Found: C, 72.32; H, 4.98, N, 0.00.
7-(4-Phenylbenzyloxy)-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM10)White crystalline solid (135 mg, 80%); melting point 160–161°C; 1H-NMR (CDCl3) δ: 3.83 (3H, s, 2′-OCH3), 5.22 (2H, s, benzyl position), 6.96 (1H, d, J = 2.5 Hz, H-8), 6.99 (1H, d, J = 8.5 Hz, H-6′), 7.02 (1H, ddd, 1H, J = 1.0 Hz, J = 8.5 Hz, H-4′), 7.09 (1H, dd, J = 2.5 Hz, J = 8.5 Hz, H-6), 7.32 (1H, dd, J = 1.5 Hz, J = 9.0 Hz, H-5′), 7.35–7.39 (2H, m, H-3′ and 4-phenylbenzyl), 7.44–7.47 (2H, m, 4-phenylbenzyl), 7.53 (2H, d, J = 8.0 Hz, 4-phenylbenzyl), 7.60–7.62 (2H, m, 4-phenylbenzyl), 7.64–7.66 (2H, m, 4-phenylbenzyl), 7.91 (1H, s, H-2), 8.22 (1H, d, J = 8.5 Hz, H-5); 13C-NMR (CDCl3) δ: 55.77 (2′-OCH3), 70.30 (benzyl position), 101.34 (C-8), 111.22 (C-6′), 114.88 (C-6), 118.71 (C-10), 120.56 (C-4′), 120.93 (C-1′), 122.65 (C-3), 127.15 (4-phenylbenzyl), 127.54 (4-phenylbenzyl), 127.96 (4-phenylbenzyl), 128.03 (4-phenylbenzyl and C-5), 128.84 (4-phenylbenzyl), 129.76 (C-3′), 131.76 (C-5′), 134.72 (4-phenylbenzyl), 140.42 (4-phenylbenzyl), 141.42 (4-phenylbenzyl), 153.81 (C-2), 157.55 (C-2′), 157.93 (C-9), 162.88 (C-7), 175.48 (–C=O); DUIS-MS (positive mode): m/z = 435 [M + H]+, 457 [M + Na]+, (negative mode): m/z = 433 [M–H]−; Anal. Calcd for C29H22O4: C, 80.17; H, 5.00; N, 0.00; O, 14.73. Found: C, 80.18; H, 4.91; N, 0.00.
7-Benzyloxy-3-(2-methoxyphenyl)-4H-chromen-4-one (ARM11)White crystalline solid (0.39 g, 79%); melting point 157–158°C; 1H-NMR (CDCl3) δ: 3.80 (3H, s, 2′-OCH3), 5.18 (2H, s, benzyl position), 6.93 (1H, d, J = 2.0 Hz, H-8), 6.98 (1H, d, J = 7.5 Hz, H-6′), 7.02 (1H, dd, J = 1.0 Hz, J = 7.5 Hz, H-4′), 7.06 (1H, dd, J = 2.0 Hz, J = 8.5 Hz, H-6), 7.32 (1H, dd, J = 2.0 Hz, J = 7.5 Hz, H-5′), 7.35–7.38 (2H, m, H-3′ and H-4″), 7.41–7.47 (4H, m, H-2″, H-3″, H-5″, and H-6″), 7.90 (1H, s, H-2), 8.21 (1H, d, J = 8.5 Hz, H-5); 13C-NMR (CDCl3) δ: 55.74 (2′-OCH3), 70.48 (benzyl position), 101.27 (C-8), 111.18 (C-6′), 114.78 (C-6), 118.64 (C-10), 120.53 (C-4′), 120.90 (C-1′), 122.62 (C-3), 127.52 (C-2″ and C-6″ or C-3″ and C-5″), 127.90 (C-5), 128.40 (C-4″), 128.77 (C-2″ and C-6″ or C-3″ and C-5″), 129.74 (C-3′), 131.74 (C-5′), 135.75 (C-1″), 153.78 (C-2), 157.51 (C-2′), 157.89 (C-9), 162.86 (C-7), 175.46 (–C=O); DUIS-MS (positive mode): m/z = 359 [M + H]+, 381 [M + Na]+, (negative mode): m/z = 357 [M–H]−; Anal. Calcd for C23H18O4: C, 77.08; H, 5.06; N, 0.00; O, 17.86. Found: C, 77.06; H, 4.80; N, 0.00.
Docking Study of pGLB1 versus ARM00Molecular docking simulations of pGLB1 (PDB ID: 3THD, chain A) to ARM00 were carried out using the Molegro Virtual Docker (version 7.0.0; Molexus, Odder, Denmark). Several preliminary steps were required before docking, such as removing water molecules, ligands, and non-targeted protein chains, and adding hydrogens. In these experiments, we used MolDock score function, which is based on a piecewise linear potential, and a re-ranking procedure was applied to the highest ranked poses to increase the docking accuracy. Affinity grid resolution was set to 0.3 Å. Ligand evaluations were based on internal energy of binding, internal H-bond formation, and Sp2–Sp2 (trigonal planar electron domain geometry) torsion angles. Candidate cavities for ligand docking were detected by including pockets with a volume of 10–10000 Å3 and a grid resolution of 0.6. Among the candidate cavities, a cavity that included DGJ was selected as an active site for docking. The cavity volume (77.2 Å3), surface (220.16 Å2), and radius (15.0 Å) were measured. The search algorithm was set to MolDock SE, which is customized with constraining poses to the cavity, after docking energy minimization and H-bond optimization. The number of runs was 15. Parameter settings were set to 1500 iterations, 50 max population size, 100 energy threshold, 300 max step, and 1.00 neighbor distance factor. All dockings were performed at 0.70 Å RMSD threshold.
Cell CultureLow-glucose Dulbecco’s modified Eagle’s medium (DMEM, D6046) was purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Fetal bovine serum (FBS, S1820) was purchased from MP Biomedicals (Solon, OH, U.S.A.). The human cervical cancer cell line, HeLa (RCB0007), was provided by RIKEN BRC through the National Bio-Resource Project of the MEXT/AMED, Japan. The HeLa cells were cultured in DMEM with 10% FBS at 37°C in 5% CO2.
Construction and Evaluation of the Cell-Based HTS SystemHeLa cells were seeded in 96-well plates (Falcon, black, 353376, Corning, NY, U.S.A.) at a density of 1 × 104 cells/well. After incubating the cells for 24 h, the medium was replaced with 259 µL of fresh medium. Subsequently, 1 µL of 3 mM N,N-dimethylsulfoxide (DMSO) solution of substrate 1 (11.5 µM final concentration) was added to each well, followed by incubation for 6 h. The medium was then removed, and the cells were washed three times with phosphate-buffered saline (PBS). After washing, 260 µL of PBS was added to each well, and fluorescence signals from each well were recorded on a multi-mode microplate reader (Spark 10M, excitation, 480/20 nm; emission, 525/20 nm; TECAN, Männedorf, Switzerland). We defined the optimal assay parameters based on the Z′ factor= 1 − (3 × SDsubstrate 1 + 3 × SDDMSO)/(Avesubstrate 1 − AveDMSO), CV (%) = (SDsubstrate 1 or DMSO/Avesubstrate 1 or DMSO) × 100, and S/B = Avesubstrate 1/AveDMSO. The inhibition rate of the compounds was calculated using the following equation: Inhibition rate (%) = (FIinhibitor candidate − FIbackground)/(FIDMSO − FIbackground) × 100.
Primary and Secondary Compound Library ScreensBoth screenings were performed using the constructed HTS system against two compound libraries, a NPDepo library (RIKEN) and a herbal medicine library (Institute of Natural Medicine, University of Toyama). For the primary screening, the final concentrations of the compounds were 1 µg/well (NPDepo library) and 9.6 µM (herbal medicine library), respectively. For the secondary screening, the varying final concentrations of the primary hit compounds were 0.25, 0.5, and 1.0 µg/well (NPDepo library), and 0.096, 0.96, and 9.6 µM (herbal medicine library), respectively. The fluorescence signals from each well were recorded using Spark 10M (excitation, 480/20 nm; emission, 525/20 nm). The inhibition rate (%) of the compounds was calculated using the above equation.
In Vitro β-Galactosidase Assays of rpGLB1Substrate 2 was prepared as separate 20 µM solutions in 50 mM sodium citrate buffer (pH 3.5 and 6.5, respectively). rpGLB1 (recombinant human β-galactosidase-1 with a C-terminal 6His tag, 6464-GH; R&D Systems, Minneapolis, MN, U.S.A.) was prepared as separate 4 ng/µL solutions in 50 mM sodium citrate buffer (pH 3.5 or 6.5). The assays were conducted by adding rpLGLB1 solution (100 µL) to each substrate solution (100 µL) followed by maintenance at 37°C in a 96-well plate (237105; Thermo Fisher Scientific, Waltham, MA, U.S.A.). The assays were followed by monitoring the FI changes of 2MeTG for 60 min at 30 s intervals using a multi-mode microplate reader (TECAN, Spark 10M, excitation, 480/20 nm; emission, 525/20 nm, optimized for 2MeTG). The % inhibition rates of the compounds were calculated using the above equation.
Third Compound Library ScreeningA third compound library screening was performed using rpGLB1 against the hit compounds from the second screen. A rpGLB1 solution (4 ng/µL, 100 µL, 50 mM sodium citrate buffer, pH 6.5), a substrate 2 solution (20 µM, 100 µL, 50 mM sodium citrate buffer, pH 6.5, containing 0.67% DMSO), and hit compound DMSO solutions (1, 2, or 4 mM; 1 µL) were added into 96-well plates, followed by incubation at 37°C in Spark 10M. The assays were followed by monitoring the FI changes for 2MeTG for 120 min at 30 s intervals using Spark 10M (excitation, 480/20 nm; emission, 525/20 nm). The inhibition rate (%) of the compounds was calculated using the above equation.
Cytotoxicity AssayHeLa cells were seeded in 96-well plates at a density of 5000 cells/well. After incubating the cells at 37°C for 24 h in 5% CO2, the medium was replaced with 100 µL of 100 µM compound fresh medium solution containing 0.5% DMSO. The cells were then incubated at 37°C for 24 h in 5% CO2. After incubation, 10 µL of Cell Counting Kit-8 solution (CK-04, Dojindo Molecular Technology, Japan) was added to each well, and the cells were incubated at 37°C for 2 h in 5% CO2. The absorbance at 450 nm of each well was measured using an iMark microplate reader (Bio-Rad, Hercules, CA, U.S.A.). All procedures were performed according to the manufacturer’s instructions and a previously reported method.20)
In Vitro Inhibition Assays for SARA rpGLB1 solution (4 ng/µL in 50 mM sodium citrate buffer, pH 6.5; 100 µL), a substrate 2 solution (20 µM in 50 mM sodium citrate buffer containing 0.67% DMSO, pH 6.5; 100 µL), and ARM00 derivatives DMSO solutions (15 mM for preliminary SAR study and 5 mM for initial SAR study; 1 µL) were added into 96-well plates, followed by incubation at 37°C in Spark 10M. The assays were followed by monitoring FI changes of 2MeTG for 120 min at 30 s intervals using Spark 10M (excitation, 480/20 nm; emission, 525/20 nm). The inhibition rate (%) of the compounds was calculated using the above equation.
Kinetic Study of Inhibition by ARM07The IC50 value was estimated from the inhibition curve, as follows. A rpGLB1 solution (4 ng/µL in 50 mM sodium citrate buffer, pH 6.5; 100 µL), a substrate 2 solution (20 µM in 50 mM sodium citrate buffer containing 0.67% DMSO, pH 6.5; 100 µL), and ARM07 DMSO solution (0.625, 1.25, or 2.5 mM) were added into 96-well plates, followed by incubation at 37°C in Spark 10 M. The assays were followed by monitoring FI changes of 2MeTG for 120 min at 30 s intervals using Spark 10M (excitation, 480/20 nm; emission, 525/20 nm). The inhibition rate (%) of the compounds was calculated using the above equation. The type of inhibition type and Ki values were determined as follows. Varying concentrations of ARM07 (1.25, 2.5, and 5.0 mM in DMSO; 1 µL), varying concentrations of substrate 2 solution 10, 20, and 30 µM in 50 mM sodium citrate buffer containing 0.67% DMSO, pH 6.5, 100 µL), and rpGLB1 solution (12 ng/µL in 50 mM sodium citrate buffer, pH 6.5; 100 µL) were added into 96-well plates, followed by incubation at 37°C in Spark 10M. The assays were followed by monitoring FI changes of 2MeTG for 120 min at 30 s intervals using Spark 10M (excitation, 480/20 nm; emission, 525/20 nm). The velocity of each reaction (FI/s/µg protein) was calculated by linear interpolation of the data. The inhibition type and the Ki value were graphically determined with a Dixon plot.
Cell-Based Inhibition AssayHeLa cells were seeded in 6-well plates at a density of 9.0 × 104 cells/well. After incubating the cells for 24 h, the medium was replaced with 1 mL of fresh medium. Subsequently, 5 µL of 10 mM ARM07 (final concentration 50 µM) or 10 or 60 mM DGJ (final concentrations of 50 or 300 µM, respectively) in DMSO were added to each well, and the cells were incubated for 2 h. The medium was removed, and the cells were washed twice with PBS, followed by immediate fixation with 10% formalin. After washing, the cells were added with 1 mL of PBS, and then the fluorescence signals of the cells were recorded using a BIOREVO BZ-9000 fluorescence microscope (Keyence, Japan) equipped with filter sets for GFP-BP (excitation, 470/40 nm; dichroic filter, 495 nm; and emission, 535/50 nm) for Alexa Fluor 488 measurements. Fluorescence images, FIs, and FAs were analyzed using BZ-analyzer ver. 2.1 (Keyence) and WinROOF 2013 ver. 1.2.0. (Mitani, Japan) according to a previously reported method.13,20)