Results and Discussion
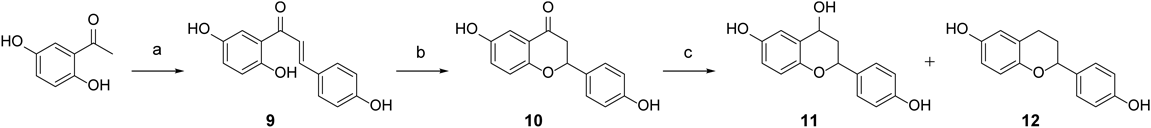
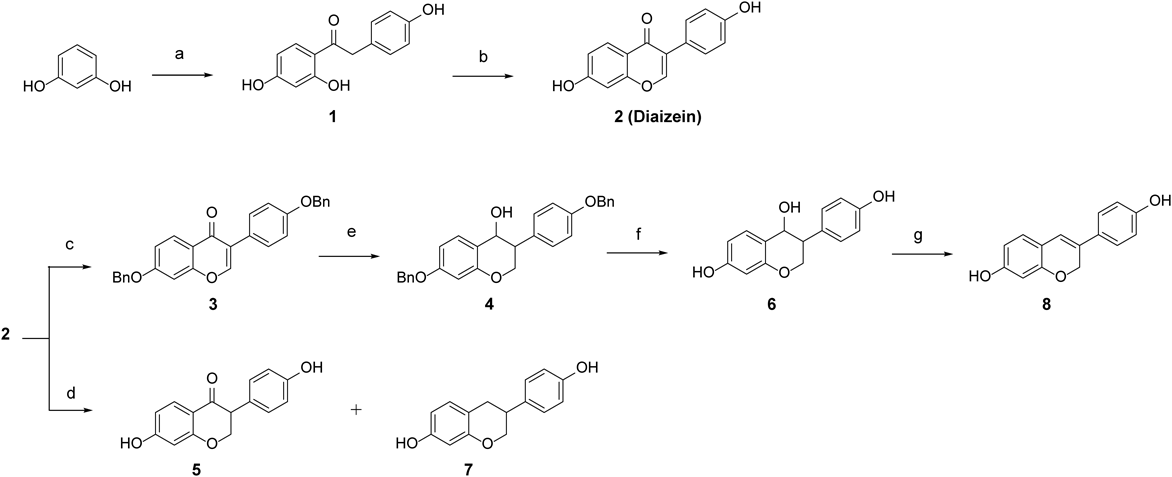
Synthesis of Isoflavonid and Flavonoid AnalogsA series of isoflavonoid analogs was prepared as described in Charts 1 and 2. Fridel–Crafts acylation of resorcinol with phenyl acetic acid in the presence of boron trifluoride diethyl etherate in N,N-dimethylformamide (DMF) yielded compound 1. Cyclization of compound 1 via treatment with N-dimethoxymethyl-N,N-dimethylamine afforded daidzein 2. Dihydrodaidzein 5 and equol 7 were obtained by palladium-catalyzed hydrogenation of daidzein 2. After protection of the hydroxyl groups of compound 2 with benzyl, reduction of ketone with NaBH4 was conducted to obtain alcohol 6. The debenzylation of compound 6 and the following elimination reaction in the acidic condition afforded dehydroequol 8. The corresponding flavonoid series were prepared as depicted in Chart 2. Condensation of 2,5-dihydroxyacetophenone with 4-hydroxybenzaldehyde in an aqueous solution of potassium hydroxide produced chalcone 9, which was cyclized to 4′,6-dihydroxyflavanone 10. Catalytic hydrogenation of flavanone 10 yielded alcohol 11 and the dehydrated compound 12.
All the prepared compounds were evaluated for their ER activities using a standard cell-based transactivation assay. Estradiol (E2) was used as a positive control for ERα and ERβ. The activity of the tested compounds was presented as the percentage of activation relative to the maximum values of the positive control. The ERα and ERβ transactivation activities of the compounds are described in Table 1.
Table 1.
In Vitro Functional Transactivation Activities of Phytoestrogen and Their Synthetic Analogs on Human ERα/β
 |
|---|
| Compounds | R1 | R2 | ER transactivation activity |
|---|
| % max (1 µM)a) | β/α ratio |
|---|
| ERα | ERβ |
|---|
| 1 | | | 9.99 | NAc) | — |
| 2 | | | 99.7 | 114 | 1.1 |
| 5 | =O | 108 | 205 | 1.9 |
| 6 | H | OH | 90.3 | 189 | 2.1 |
| 7 | H | H | 208 | 334 | 1.6 |
| 8 | — | H | 187 | 335 | 1.8 |
| 9 | | | 83.1 | 93.3 | 1.1 |
| 10 | =O | 119 | 108 | 0.9 |
| 11 | H | OH | 37.1 | 82.3 | 2.2 |
| 12 | H | H | 100 | 110 | 1.1 |
| E2b) | | | 100 | 100 | 1.0 |
a) Fold activation relative to maximum activation obtained with E2 (20 nM) for ERα and β. The compounds were tested in at least three separate experiments. b) The concentration of E2 for transactivation activity is 20 nM. c) NA means not active, which is for compounds producing ER transactivation activity lower than 10% at 1 µM.
Several tested isoflavonoid and flavonoid analogs showed a higher ER activity than that of daidzein. At first, the effect of C-ring of isoflavonids and flavonoids on the activity and selectivity was examined. C-ring was quite essential for ER activity. Compound 1 had no ER activity and chalcone 9 also revealed a lower ER activity than the most cyclized compounds. Isoflavone (2, daidzein) was transformed to isoflavanone (5), isoflavanol (6), isoflavane (7), or isoflavene (8) scaffolds via reduction of C-ring and/or reduction of C4 ketone. These modification of C-ring mostly increased ERβ activity and selectivity over ERα. Isoflavanone 5 exhibited a 1.9-fold higher ERβ activity compared to daidzein 2 and similar ERα activity. The C4-ketone of compound 5 was further reduced to a 4-hydroxyl group (6) to investigate whether different oxidation states affect ER activity. Isoflavanol (6) had slightly lower ERα and ERβ activities than isoflavanone (5), but a higher selectivity for ERβ (2.1-fold) over ERα. Among these isoflavonoids, equol 7 and dehydroequol 8 were the most potent, with 1.6 and 1.8-fold ERβ selectivities, respectively. It implied that 4-hydroxyl or ketone groups on the C-ring was not essential for ER activity.
The ER activities of the flavonoid analogs (10–12) were compared to those of the corresponding isoflavonoids. Most of the tested flavonoids revealed a lower activity and selectivity than the corresponding isoflavonoids. The ERβ activities of compounds 10, 11, and 12 were less than half of those of the respective isoflavonoids: 5, 6, and 7. Flavanone 10 and flavane 12 exhibited a loss of ERβ selectivity. Reduction of ketone at the C4 of compound 10 to a hydroxyl group (11) decreased ERα/β activities, but retained ERβ selectivity, as observed in isoflavonoids. Overall, the results indicated that the ER activity and selectivity of isoflavonoid scaffolds are more favorable than those of flavonoids. In addition, the ER activity of isoflavonoids are highly influenced by C-ring modification.
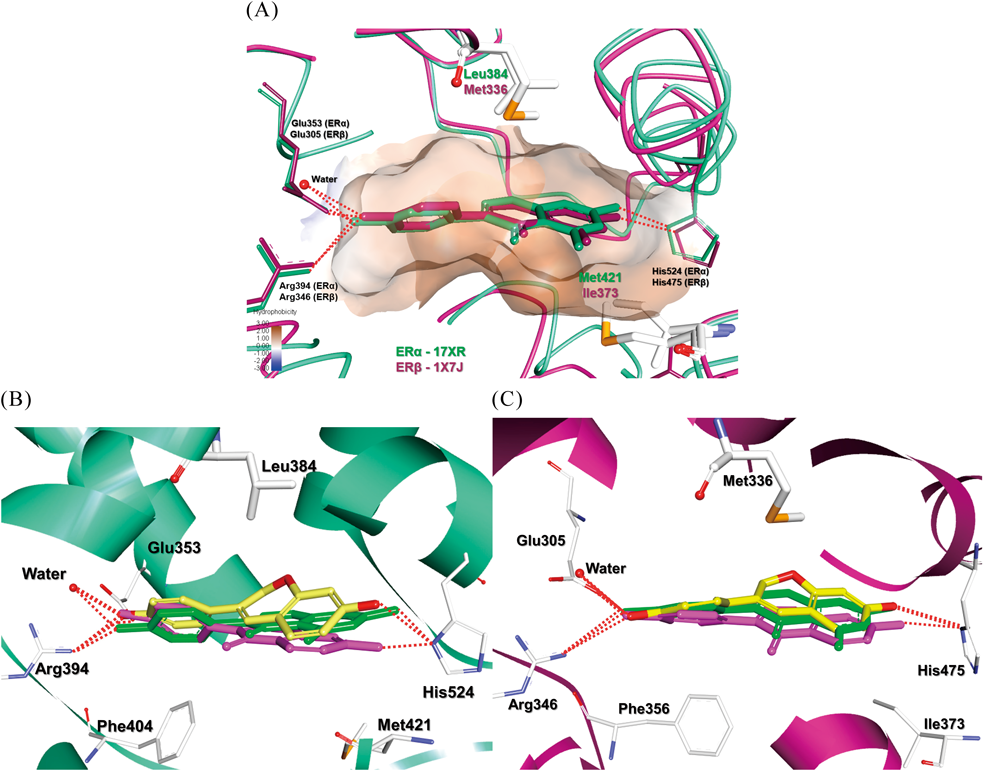
Molecular Docking StudyDocking analysis and visualization were performed using SYBYL-X version 2.1.1 and Discovery Studio Visualizer. Crystal structures of ERα and ERβ in complex with genistein (PDB code: ERα -17XR, ERβ -17XJ) were employed for the docking study and internal default parameters of Suflex Dock were used for all the variables.
The structure of ligand binding domain (LBD) of ERα and ERβ is highly conserved, and the differences within the ligand binding cavity are only at the two amino acid positions. The two characteristic residues for each subtype are ERα Leu384 vs. ERβ Met336 and ERα Met421 vs. ERβ Ile373. The two paired amino acids within the LBD of ERα and ERβ lead to differences in their structures and contribute to ligand selectivity (Fig. 3A).
Initially, the crystal structure of genistein complexed with ERs was thoroughly analyzed.11) It has been reported that the γ-pyrone ring centroid of genistein is approximately 4.0–4.5 Å from ERβ Met336 Cε atom and 6.2 Å from ERα Leu384 Cδ1 atom. Also, the carbon at 3-position of the C-ring was about 4.2–4.6 Å from ERβ Met336 Sδ atom, and 4.8 Å from ERα Leu384 Cδ2 atom. Thus, ERβ Met336 clearly has a greater potential to achieve attractive interactions with C-ring of genistein than ERα Leu384.
We investigated binding modes of daidzein at the active site of ERs in comparison with crystal structure of genistein. Daidzein 2 fits well into the active site of ERs and its two hydroxyl groups are involved in an important hydrogen bonding network. However, unlike genistein, daidzein was less favorable for the hydrophobic interaction with Met336 of ERβ. In fact, the γ-pyrone ring of daidzein 2 centroid was about 4.8–5.1 Å from the ERβ Met336 Cε atom further away than that of genistein, which may explain the loss of ERβ selectivity.
Next, we examined closely binding modes of isoflavonids (5–8) and flavonoids (10–12) at the active site of ERs in comparison with that of daidzein 2. The A and B-ring hydroxyl groups of the most isoflavonoids were appropriately positioned for critical polar interactions with His524 and Arg394-Glu353-water in ERα or the corresponding His475 and Arg346-Glu305-water in ERβ (Figs. 3B, C), respectively. On the other hand, all flavonoids showed a loss in some of the major hydrogen bonding interactions (ERα-His524, ERβ-His475) at the active sites of both ERs.
Reduction of C-ring and/or C4 ketone in isoflavonids altered the conformation of the C-rings, causing them to be out-of-plane form. This change of the C ring in the folded form appears to lead to a better hydrophobic interaction with the characteristic residues ERβ Met336 and ERα Leu384.
Dehydroequol 8, where the γ-pyrone ring of daidzein 2 was reduced and deoxygenated, increased ERβ selectivity by about 1.8 times. Similar to genistein, dehydroequol 8 was well positioned for van der Waals interaction of the pyran ring with Met336 in ERβ. The pyran ring centroid was approximately 3.6–4.2 Å from ERβ Met336 Cε atom and 4.2–4.6 Å from ERα Leu384 Cδ1 atom. This may contribute to the ERβ selectivity of dehydroequol 8. In contrast, neither ERα Met421 and ERβ-Ile373 made a significant contribution to the ERβ selectivity.
Consequently, the docking studies have clearly shown that out-of-plane C-rings of isoflavonoids could bring favorable hydrophobic interactions with the characteristic residues of ERs. As a result, compound 8 was capable of presenting ERβ selectivity along with ERα activity.
In Vitro Anti-neuroinflammatory ActivitySubsequently, the neuroprotective effects of these compounds were examined because some phytoestrogens and their metabolites are known to have anti-neuroinflammatory effects in an ER-dependent manner. The anti-neuroinflammatory activities of the synthesized compounds were evaluated by measuring inhibition of NO production in LPS-stimulated BV-2 microglial cells. Cell viability was determined after cell exposure to LPS in the presence or absence of tested compounds. None of the tested compounds showed cytotoxicity at 20 µM. The anti-neuroinflammatory activity of the tested compounds and cell viability results are described in Table 2.
Table 2. Anti-neuroinflammatory Effects of Phytoestrogen and Their Synthetic Analogs against LPS-Induced Neurotoxicity in BV-2 Microglial Cells
| Compounds | IC50a) (µM) | Cell viabilityb) (%) |
|---|
| 1 | >500 | 104 ± 14 |
| 2 | 118 | 107 ± 3.9 |
| 5 | 32.1 | 128 ± 13 |
| 6 | 5.50 | 133 ± 7.2 |
| 7 | 2.40 | 94.5 ± 14 |
| 8 | 2.80 | 133 ± 5.7 |
| 9 | 2.90 | 97.3 ± 9.1 |
| 10 | 7.60 | 127 ±11 |
| 11 | 33.7 | 103 ± 8.8 |
| 12 | 5.50 | 113 ± 8.8 |
| L-NMMAc) | 16.6 | 127 ± 8.4 |
a) IC50 value of each compound was defined as the concentration (µM) that caused 50% inhibition of NO production in LPS-activated BV-2 cells. b) Cell viability after treatment with 20 µM of each compound was determined by an MTT assay and is expressed as percentage of LPS (%). The results are averages of three independent experiments, and the data are expressed as mean ± standard deviation (S.D.) c) L-NMMA (NG-methyl-L-arginine) is a positive control.
In summary, a small set of isoflavonoid and flavonoid analogs were prepared and evaluated for ER transactivation activity and inhibitory activity on NO production in LPS-stimulated microglia. Our SAR study indicated that saturation of C-ring mostly enhanced ERβ activity and selectivity in the series of isoflavonoids but not in flavonoids. Overall, isoflavone scaffold was favorable for ERβ selectivity. Among the tested compounds, the well-known daidzein metabolites equol 7 and dehydroequol 8 showed the most potent ER transactivation and neuroprotective activity. Our study showed that an improvement in the selectivity of isoflavonoids for ERβ could be achievable with a minor tuning of C-ring conformation. These results provide insights into the development of useful ERβ-selective therapeutic agents for treating inflammatory-mediated neurodegenerative diseases.
Experimental
ChemistryMost of the reagents and solvents used were purchased from Aldrich Chemicals and used without further purification, with the following exceptions. Tetrahydrofuran (THF) was distilled from sodium benzophenone ketyl. Acetonitrile, methylene chloride, triethylamine (Et3N), pyridine, and dimethyl formamide (DMF) were distilled from calcium hydride under nitrogen atmosphere. Column chromatography was performed using a silica gel 60 (230–400 mesh, Merck) with the indicated solvents. TLC was performed using Kieselgel 60 F254 plates (Merck). IR spectra were recorded on a JASCO FT/IR 430 spectrophotometer. 1H- and 13C-NMR spectra were recorded in CDCl3 and, CD3OD or dimethyl sulfoxide (DMSO)-d6 on a Bruker Avance III HD 500 NMR spectrometer. Chemical shifts (δ) are expressed in parts per million (ppm) downfield from an internal standard, tetramethylsilane.
1-(2,4-Dihydroxyphenyl)-2-(4-hydroxyphenyl)ethanone 1Resorcinol (1.0 g, 9.08 mmol) and 4-hydroxyphenylacetic acid (1.4 g, 9.08 mmol) in distilled DMF (5 mL) was heated at 70 °C for 30 min. The reaction mixture was cooled to 0 °C, and then added dropwise sloswly to boron trifluoride etherate (20 mL) and stirred at room temperature for 4 h. The reaction mixture was stirred until the starting material was completely consumed in TLC and quenched with sodium bicarbonate solution. Afterward, it was basified by 1 N NaOH (pH = 8) and extracted with ethyl acetate. The extracts were dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate = 2 : 1) to afford compound 1 (1.9 g, 7.89 mmol, 86%) as a yellow solid. Rf = 0.32 (n-hexane/EtOAc = 2 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.85 (1H, d, J = 8.8 Hz), 7.10 (1H, d, J = 8.4 Hz), 6.73 (2H, d, J = 8.4 Hz), 6.35 (1H, dd, J = 9.2, 2.4 Hz), 6.24 (1H, d, J = 8.8, 2.4 Hz); 13C-NMR (100 MHz, CD3OD) δ: 174.5, 172.6, 170.3, 162.1, 154.2, 153.5, 136.2, 134.1, 127.5, 112.4, 112.3, 106.3, 103.4, 40.3.
7-Hydroxy-3-(4-hydroxyphenyl)-4H-1-benzopyran-4-one 2To a solution of compound 1 (1.9 g, 0.74 mmol) in DMF (2 mL) was added N,N-dimethylformamide dimethyl etherate and stirred at room temperature for 4 h. Subsequently, 1 N HCl solution was added and the mixture was stirred at room temperature for 6 h. The reaction mixture was filtered and washed with ether, and the filtrate was purified by silica gel column chromatography (eluent: n-hexane/EtOAc = 1 : 1) to afford compound 2 (1.5 g, 0.58 mmol, 74%) as a yellow solid. Rf = 0.28 (n-hexane/EtOAc = 1 : 1); 1H-NMR (400 MHz, DMSO) δ: 10.75 (OH, s), 9.55 (OH, s), 8.29 (1H, s), 7.96 (1H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz), 6.93 (1H, dd, J = 8.8, 2.0 Hz), 6.85–6.79 (3H, m); 13C-NMR (100 MHz, DMSO) δ: 184.2, 172.1, 166.9, 166.7, 162.3, 139.6, 136.8, 132.9, 132.0, 124.6, 124.4, 114.1, 111.6.
7-Benzyloxy-3-(4-benzyloxyphenyl)chrome-4-one 3Compound 2 (540.8 mg, 2.11 mmol) was dissolved in dry DMF (2 mL), and K2CO3 (961.8 mg, 6.38 mmol) and benzyl bromide (1113.4 mg, 6.38 mmol) were added. The mixture was heated at 40 °C for 2 h. After cooling the mixture to room temperature, ethyl acetate and water were added. The organic layer was washed several times with water and brine, and then dried over MgSO4. The residue was purified by silica gel column chromatography (eluent; n-hexane/EtOAc = 4 : 1) to afford compound 3 (743.2 mg, 1.70 mmol, 79%) as a yellow solid. Rf = 0.27 (n-hexane/EtOAc = 4 : 1); 1H-NMR (400 MHz, CDCl3) δ: 8.22 (1H, d, J = 9.2 Hz), 7.91 (1H, s), 7.50–7.33 (10H, m), 7.07–7.03 (3H, m), 6.93 (1H, s), 5.18 (2H, s), 5.11 (2H, s); 13C-NMR (100 MHz, CDCl3) δ: 175.8, 162.9, 152.1, 135.7, 130.1, 128.8, 128.6, 128.4, 127.9, 127.5, 127.4, 115.0, 114.9, 105.0, 101.3, 70.5, 70.1.
4′,7-Dibenzyloxyisoflavan-4-ol 4NaBH4 (151.0 mg, 4.00 mmol) was added to a stirred solution of 3 (743.2 mg, 1.70 mmol) in distilled THF (20 mL) at 0 °C under argon. The mixed solution was stirred at 0 °C for 10 min, warmed to room temperature, and stirred overnight. Next, the mixture was extracted with ethyl acetate, washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent; chloroform/n-hexane/ethyl acetate = 20 : 5 : 1) to afford compound 4 (310.8 mg, 0.70 mmol, 41%). Rf = 0.25 (chloroform/n-hexane/EtOAc = 20 : 5 : 1); 1H-NMR (400 MHz, CDCl3) δ: 7.30 (5H, m), 7.21 (3H, d, J = 8.8 Hz), 7.12 (1H, d, J = 8.4 Hz), 6.82 (2H, d, J = 8.8 Hz), 6.51 (1H, dd, J = 8.4, 2.4 Hz), 4.57 (1H, d, J = 4.8 Hz), 4.58 (2H, s), 4.49 (1H, dd, J = 11.2, 3.2 Hz), 4.34 (1H, dd, J = 11.2, 4.8 Hz), 3.78 (3H, s), 3.76 (3H, s), 3.34 (1H, m); 13C-NMR (100 MHz, CDCl3) δ: 159.8, 157.8, 154.9, 136.5, 152.9, 143.3, 139.2, 124.5, 121.1, 120.0, 109.4, 102.8, 101.1, 100.7, 95.3, 70.6, 71.0, 39.3.
7-Hydroxy-3-(4-hydroxyphenyl)-2,3-dihydrochromen-4-one 5Acetic acid (2 mL) and 10% Pd (100 mg, 100 wt%) was added to a solution of 2 (100 mg, 0.39 mmol) in EtOH (18 mL) at room temperature. The resulting mixture was stirred at room temperature for 8 h. After completion of reaction, the mixture was filtered through a celite pad and washed with methanol. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent, n-hexane/EtOAc = 1 : 1) to afford compound 5 (30.7 mg, 0.12 mmol, 30%) as a red solid: Rf = 0.24 (n-hexane/EtOAc = 1 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.74 (1H, d, J = 8.8 Hz), 7.07 (2H, d, J = 8.4 Hz), 6.74 (2H, d, J = 8.8 Hz), 6.54 (1H, dd, J = 8.8, 2.0 Hz), 6.33 (1H, d, J = 2.4 Hz); 13C-NMR (100 MHz, CD3OD) δ: 172.3, 170.6, 163.1, 157.9, 156.2, 131.1, 130.9, 127.3, 110.9, 112.1, 106.5, 103.1, 67.3, 53.2.
4′,7-Dihydroxyisoflavan-4-ol 6The substrate dibenzyl compounds 3 (196.2 mg, 0.45 mmol), ammonium formate (567.5 mg, 9.0 mmol), 20% Pd(OH)2 (196.2 mg, 100 wt%) and THF/EtOH/H2O (20 mL) were placed in a 50-mL reactor. The reaction mixture was stirred for 15-20 min at room temperature under nitrogen. After the reaction was complete (monitored by TLC) the catalyst was removed by filtration and washed with 5 mL of methanol. The crude product was purified by flash chromatography (eluent: n-hexane/EtOAc = 1 : 1) to produce pure 6 (73 mg, 0.28 mmol, 62%) as a yellow oil. Rf = 0.18(n-hexane/EtOAc = 1 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.18 (1H, d, J = 8.4 Hz), 7.03 (2H, d, J = 8.8 Hz), 6.71 (2H, d, J = 8.8 Hz), 6.38 (1H; dd; J = 8.4, 2.4 Hz), 6.25 (1H, d, 2.4 Hz), 4.74 (1H, d, J = 2.4 Hz), 4.24 (1H, dd, J = 11.6, 3.2 Hz), 4.14 (1H; dd; J = 10.0, 8,8 Hz), 3.01–2.96 (1H, m); 13C-NMR (100 MHz, CD3OD) δ: 159.1, 157.4, 156.8, 131.9, 130.9, 130.1, 117.9, 116.3, 109.6, 103.3, 69.9, 69.3, 47.7.
3,4-Dihydro-3-(4-hydroxyphenyl)-2H-1-benzopyran-7-ol 7The title compound was prepared from 2 using the same synthesis procedure as that of 5. The crude product was purified by flash chromatography (MeOH/CHCl3 = 1 : 1) to produce pure 7 (43.5 mg, 0.18 mmol, 46%) as a yellow oil. Rf = 0.24 (MeOH/CHCl3 = 2 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.07 (2H, d, J = 8.8 Hz), 6.84 (1H, d, J = 8.4 Hz), 6.74 (2H, d, J = 8.8 Hz), 6.30 (1H; dd; J = 8.0, 2.4 Hz), 6.33 (1H, d, J = 2.4 Hz), 4.18 (1H; t; J = 10.4, 3.6, 1.6 Hz), 3.88 (1H; dd; J = 10.4, 3.6 Hz), 3.07 (1H, m), 2.85 (2H, m); 13C-NMR (100 MHz, CD3OD) δ: 170.8, 170.6, 161.9, 153.2, 155.7, 130.2, 129.9, 126.3, 115.7, 111.0, 105.3, 102.1, 65.3, 53.2, 41.2.
3-(4-Hydroxyphenyl)-2H-chromen-7-ol 8To a solution of compound 6 (61.9 mg, 0.24 mmol) in methanol was added 1 N HCl. The mixture was then stirred at room temperature for 1 h. Afterward, the reaction mixture was diluted and extracted with EtOAc. The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: n-hexane/EtOAc = 4 : 1) to afford compound 8 (23.0 mg, 0.12 mmol, 49%) as a yellow oil. Rf = 0.24 (n-hexane/EtOAc = 1 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.27 (2H, d, J = 8.0 Hz), 6.94 (1H, d, J = 8.0 Hz), 6.79 (1H, d, J = 8.0 Hz), 6.61 (1H, s), 6.38 (1H; dd; J = 12.0, 2.4 Hz), 6.25 (1H, d, J = 2.4 Hz); 13C-NMR (100 MHz, CD3OD) δ: 185.6, 173.4, 171.5, 165.2, 158.1, 156.1, 132.2, 131.1, 123.9, 115.2, 112.3, 107.9, 102.4, 43.5
1-(2,5-Dihydroxyphenyl)-3-(4-hydroxyphenyl)-propenone 9KOH (60%, 8 mL) was added to a solution of 5-hydroxy-2-methyl-benzoic acid (1 g, 6.57 mmol) in EtOH (1 mL) and the mixture was then stirred at room temperature for 30 min. 4-Hydroxybenzaldehyde (785.6 mg, 6.40 mmol) was added to the reaction mixture, which was then stirred at 100 °C for 2 h. The reaction mixture was cooled and quenched with ice water (6 mL), acidified to pH = 5 by the addition of aqueous HCl solution (2.6 mL). Afterward, the reaction mixture was washed with water (25 mL) and filtrated to afford compound 9 (986.0 mg, 3.84 mmol, 57%) as a yellow solid. Rf = 0.35 (methanol/chloroform = 2 : 1); 1H-NMR (400 MHz, CD3OD) 7.85 (1H, d, J = 15.6 Hz), 7.64 (2H, d, J = 8.4 Hz), 7.59 (1H, d, J = 15.6 Hz), 7.42 (1H, d, J = 2.4 Hz), 7.03 (1H; dd; J = 8.8, 2.4 Hz), 6.86 (2H, d, J = 8.4 Hz), 6.82 (1H, d, J = 8.8 Hz); 13C-NMR (100 MHz, CD3OD) δ: 195.1,161.9, 157.6, 150.6, 146.9, 132,0, 127.6, 125.5, 119.6, 118.2, 117.0, 115.4.
6-Hydroxy-2-(4-hyroxyphenyl)chroman-4-one 10A solution of compound 9 (986.0 mg, 3.84 mmol) in MeOH (4 mL) was reacted with concentrated HCl solution (3 mL) at 100 °C overnight. The reaction mixture was quenched with water, and MeOH was removed under vacuum. The residue was then filtered with water to afford compound 10 (485.0 mg, 1.97 mmol, 59%). Rf = 0.35 (n-hexane/EtOAc = 2 : 1); IR (neat, cm−1) 3377, 1656, 1601, 1516, 1465, 1234, 1163; 1H-NMR (400 MHz, CDCl3) δ: 7.71 (1H, d, J = 8.7 Hz), 7.38 (2H, d, J = 8.7 Hz), 6.41 (1H, d, J = 2.3 Hz), 5.44 (1H; dd; J = 13.0, 3.0 Hz), 3.03 (1H; dd; J = 16.8, 13.0 Hz), 2.66 (1H; dd; J = 16.8, 3.0 Hz); 13C-NMR (100 MHz, CDCl3) δ: 189.7, 168.6, 168.1, 162.2, 156.7, 151.1, 136.5, 127.7, 127.5, 121.9, 118.7, 115.6, 111.0, 79.4, 43.7, 20.0, 19.9.
2-(4-Hydroxyphenyl)chroman-4,6-diol 11Acetic acid (10.3 mL), 10% palladium (205.7 mg, 80 wt%) was added to a solution of compound 10 (257.1 mg, 1.00 mmol) in distilled EtOH (25.7 mL). The mixture was stirred at room temperature for 8 h and filtered through a celite pad. Next, the filtrate was washed with methanol, and the residue was purified by silica gel column chromatography (eluent; n-hexane/ethyl acetate = 6 : 1 v/v) to afford compound 11 (91 mg, 0.35 mmol, 35%). Rf = 0.23 (n-hexane/EtOAc = 6 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.27 (2H, d, J = 8.4 Hz), 6.95 (1H, s), 6.80 (1H, d, J = 8.4 Hz), 6.63–6.58 (2H, m), 5.00–4.97 (2H, m), 2.32 (1H; ddd; J = 12.8, 6.4, 6.0 Hz), 2.09–1.98 (1H, m); 13C-NMR (100 MHz, CD3OD) δ: 150.6, 147.9, 132.1, 127.2, 126.7, 121.5, 116.4, 115.3, 114.7, 112.7, 76.7, 65.2, 39.7
2-(4-Hydroxyphenyl)chroman-6-ol 12The title compound was prepared from 10 using the same synthesis procedure as that of 11. The crude product was purified by flash chromatography (eluent: methanol/chloroform = 1 : 1 v/v) to produce pure 12 (111.8 mg, 0.46 mmol, 46%). Rf = 0.23 (methanol/chloroform = 1 : 1); 1H-NMR (400 MHz, CD3OD) δ: 7.23 (2H; d; J = 6.4, 1.6 Hz), 6.75 (1H, d, J = 8.0 Hz), 6.58 (1H, d, J = 2.8 Hz), 6.51 (1H, m), 4.83 (1H, d, J = 2.4 Hz), 2.93 (1H; ddd; J = 12.0, 8.0, 4.0 Hz), 2.50 (1H, m) 2.09 (1H, m), 1.31 (1H, m); 13C-NMR (100 MHz, CD3OD) δ: 156.7, 148.4, 127.1, 122.3, 116.7, 114.9, 114.7, 113.8, 77.5, 29.8, 25.0.
Molecular Docking StudyIn silico docking of tested compounds with the 3D coordinates of the X-ray crystal structures of the estrogen receptors ERα and ERβ (PDB code; 1X7R and 1X7J) was conducted using the SYBYL-X program version 2.1.1. In this automated docking program, the flexibility of the ligands is considered while the protein or biomolecule is considered as a rigid structure. The ligand is built in an incremental fashion, where each new fragment is added in all possible positions and conformations to a pre-placed base fragment inside the active site. The protomol was generated using the crystallized ligand with a threshold of 0.50 as the default setting. All molecules used for docking were sketched in the SYBYL, and energy minimization was performed using the Tripos force field and Gasteiger–Huckel charge with 20000 iterations of conjugate gradient method with a convergence criterion of 0.05 kcal/mol. All other parameters were at their default settings
ERE-luciferase Reporter Gene AssayCV-1 cells were seeded in 48-well plate at a density of 1.5 × 105 cells/well. One day after seeding, the cells were co-transfected with 30 ng hERα (or hERβ) and 100 ng pERE-luciferae plasmid in each well for 6 h, and further treated with vehicle and samples for 24 h before harvesting for the luciferase assay. To normalize the transfection efficiency, β-galactosidase plasmid was co-transfected. The luciferase activities were measured using luciferase assay system (Promega Corp., Madison, WI, U.S.A) and the β-galactosidase activities were measured as the absorbance at 410 nm by using an enzyme-linked immunosorbent assay (ELISA) plate reader. Data are reported as relative luciferase activity divided by the β-galactosidase activity.
Nitrite AssayNitric oxide (NO) was measured in BV2 murine microglia cells with or without activation with lipopolysaccharide. BV2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) 10% fetal bovine serum and 1% antibiotic penicillin and streptomycin (PS). Cells were seeded in the 96 well plate with the density of 4 × 104 cells/well and incubated for 24 h. Seeded cells were first treated with compound and after 30 min 100 ng/mL LPS activation was performed and incubated for 24 h in the incubator maintaining 5% CO2 and 37 °C temperature. Fifty microliters of treated cell supernatant was transferred to new plate and mixed with equal volume of Gries reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% N-1-napthylethylenediamine dihydrochloride in 1 : 1 proportion). The pink color appearance in the mixture due to the presence of nitrite was compared using NaNO2 as a standard and the absorbance was measured at 540 nm on a microplate reader. NG-Monomethyl-L-arginine (L-NMMA) was used as a positive control.
Cell Viability AssayCytotoxicity of the prepared compounds was analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. BV2 cells were seeded in the 96 well plate, treated with compounds, and activated with LPS for 24 h. After this treatment, conditioned medium was used for NO assay and the cells were incubated with 0.5% MTT solution for about 1 h in the incubator and after the cells stained blue, MTT solution were suction out and 200 µL of DMSO has been added. Addition of DMSO yield purple color formazan solution and its optical density was measured at 570 nm on microplate reader