Experimental
SynthesisAll reactions were carried out under a nitrogen atmosphere. Column chromatography was performed on Kanto Chemical silica gel 60 N (spherical, neutral, 40–50 µm). The purity and retention time (tR) of all final compounds were measured using a JASCO HPLC system equipped with a TSKgel ODS-120H column (4.6 mm I.D. × 15 cm, 5 µm) and a UV detector operated at 220 nm. Gradient elution was performed at a flow rate of 1.0 mL/min with a mixture of acetonitrile solvent and 0.1% trifluoroacetic acid in water. 1H-NMR spectra were measured on a JEOL JNM-ECZ400S (400 MHz) or JNM-ECA500WB (500 MHz). High resolution (HR)MS were recorded on a JEOL JMS-SX 102 A mass spectrometer.
(Z)-1,1,1-Trifluoro-4-hydroxy-4-(p-tolyl)-3-buten-2-one (1)4′-Methylacetophenone (100 µL, 753 µmol) was dissolved in dry tetrahydrofuran (THF) (2.0 mL), and 60% sodium hydride (NaH) (65 mg, 1.63 mmol, 2.2 equivalent (equiv.)) was added under ice-cold. After stirring for 1 h under 0 °C, ethyl trifluoroacetate (135 µL, 1.13 mmol, 1.5 equiv.) was added, and the reaction mixture was stirred for 3.5 h at room temperature. The reaction mixture was evaporated, added ice–water (5 mL), acidified to pH 6 with 1 N hydrochloric acid (HCl), and extracted 3 times with ethyl acetate (EtOAc). The organic layer was washed with water, dried over magnesium sulfate (MgSO4), filtered, and concentrated. The residue was washed with n-hexane, and evaporated. The solid was dissolved in dichloromethane (CH2Cl2), and evaporated to afford 1 (140 mg, 608 µmol, 81%). 1H-NMR (400 MHz, CDCl3) δ: 7.86 (2H, d, J = 8.4 Hz), 7.31 (2H, d, J = 8.4 Hz), 6.55 (1H, s), 2.45 (3H, s).
4-(5-(p-Tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic Acid (2)4-Hydrazinobenzoic acid (361 mg, 2.37 mmol, 1.1 equiv.) was dissolved in dry ethanol (EtOH) (30 mL), added 2 N HCl (1.3 mL) and 1 (503 mg, 2.19 mol). After stirring for 0.5 h at 75 °C, the reaction mixture was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated to afford 2 (741 mg, 2.14 mmol, 98%). 1H-NMR (400 MHz, (CD3)2CO) δ: 8.10 (2H, d, J = 8.8 Hz), 7.51 (2H, d, J = 8.8 Hz), 7.24 (4H, s), 7.00 (1H, s), 2.35 (3H, s).
(4-(5-(p-Tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)phenyl)methanol (3)Lithium aluminum hydride in THF (200 µL, 200 µmol, 1.4 equiv.) was slowly added to dry THF (500 µL), and 2 (in 1.5 mL THF) (51 mg, 147 µmol) was added under ice-cold. After stirring for 0.5 h at 0 °C, the reaction mixture was stirred for 1.5 h at room temperature. The reaction mixture was quenched with reverse osmosis (RO) water and 15% sodium hydroxide (NaOH) aq., filtered with celite, and concentrated. The residue was purified by silica gel column chromatography (CH2Cl2 only) to afford 3 (37 mg, 111 µmol, 76%). Purity: 96.4%; tR: 29.1 min; 1H-NMR (400 MHz, CDCl3) δ: 7.36 (2H, d, J = 8.0 Hz), 7.31 (2H, d, J = 8.8 Hz), 7.12 (4H, q, J = 8.8, 12.4 Hz), 6.72 (1H, s), 4.73 (2H, d, J = 6.0 Hz), 2.35 (3H, s); HRMS (electrospray ionization-time-of-flight (ESI-TOF)) m/z [M + H]+: 333.1196 (Calcd for C18H16F3N2O: 333.1215).
1-(4-(Methoxymethyl)phenyl)-5-(p-tolyl)-3-(trifluoromethyl)-1H-pyrazole (4)Sixty percent NaH (23 mg, 575 µmol, 3.3 equiv.) was dissolved in dry THF (2.0 mL), and 3 (in 2.0 mL THF) (57 mg, 172 µmol) was added on ice bath. After stirring for 1.5 h on ice bath, methyl iodide (32.2 µL, 517 µmol, 3.0 equiv.) was added. After stirring for 19 h at room temperature, the reaction mixture was quenched with DI water (1.0 mL), extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc = 10 : 1) to afford 4 (38 mg, 110 µmol, 64%). Purity: 96.3%; tR: 32.5 min; 1H-NMR (400 MHz, CDCl3) δ: 7.31 (4H, q, J = 8.4, 15.6 Hz), 7.11 (4H, q, J = 8.8, 10.4 Hz), 6.71 (1H, s), 4.47 (2H, s), 3.40 (3H, s), 2.35 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 347.1387 (Calcd for C19H18F3N2O: 347.1371).
Ethyl 4-(5-(p-Tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (5)2 (50 mg, 144 µmol) was dissolved in dry EtOH, and added SOCl2 (52.0 µL, 721 µmol, 5.0 equiv.) under ice-cold. After stirring for 24 h at room temperature, the reaction mixture was quenched with saturated sodium hydrogen carbonate (NaHCO3) aq. (1.5 mL), and evaporated. The residue was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc = 10 : 1) to afford 5 (20 mg, 53.4 µmol, 37%). Purity: 96.8%; tR: 34.7 min; 1H-NMR (400 MHz, CDCl3) δ: 8.04 (2H, d, J = 7.2 Hz), 7.39 (2H, d, J = 7.2 Hz), 7.13 (4H, q, J = 8.0, 19.2 Hz), 6.73 (1H, s), 4.38 (2H, q, J = 7.2, 14.4 Hz), 2.37 (3H, s), 1.40 (3H, t, J = 7.2 Hz); HRMS (ESI-TOF) m/z [M + H]+: 375.1328 (Calcd for C20H18F3N2O2: 375.1320).
Propyl 4-(5-(p-Tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (6)The title compound was synthesized from 2 (51 mg, 147 µmol) in 68% yield using the method described for the preparation of 5. 1-Propanol was used as a solvent instead of EtOH. Purity: 99.0%; tR: 35.8 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.8 Hz), 7.39 (2H, d, J = 8.8 Hz), 7.13 (4H, q, J = 8.4, 20.4 Hz), 6.73 (1H, s), 4.28 (2H, t, J = 7.2 Hz), 2.37 (3H, s), 1.85–1.75 (2H, m), 1.03 (3H, t, J = 7.2 Hz); HRMS (ESI-TOF) m/z [M + H]+: 389.1481 (Calcd for C21H20F3N2O2: 389.1477).
Isopropyl 4-(5-(p-Tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (7)The title compound was synthesized from 2 (52 mg, 150 µmol) in 22% yield using the method described for the preparation of 5. 2-Propanol was used as a solvent instead of EtOH. Purity: 95.1%; tR: 35.8 min; 1H-NMR (400 MHz, CDCl3) δ: 8.02 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz), 7.12 (4H, q, J = 8.0, 20.8 Hz), 6.73 (1H, s), 5.30–5.20 (1H, m), 2.37 (3H, s), 1.37 (6H, d, J = 6.4 Hz); HRMS (ESI-TOF) m/z [M + H]+: 389.1484 (Calcd for C21H20F3N2O2: 389.1477).
tert-Butyl 4-(5-(p-Tolyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (8)2 (50 mg, 144 µmol), benzyltriethylammonium chloride (36 mg, 158 µmol, 1.1 equiv.) and potassium carbonate (K2CO3) (496 mg, 3.59 mmol, 25 equiv.) were dissolved in N,N-dimethylacetamide (1.0 mL), and added 2-bromo-2-methylpropane (723 µL, 6.48 mmol, 45 equiv.). After stirring for 19 h at 55 °C, the reaction mixture was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc = 4 : 1) to afford 8 (52 mg, 129 µmol, 90%). Purity: 96.2%; tR: 37.0 min; 1H-NMR (400 MHz, CD3OD) δ: 7.99 (2H, d, J = 8.0 Hz), 7.40 (2H, d, J = 8.8 Hz), 7.18 (4H, q, J = 8.0, 18.4 Hz), 6.91 (1H, s), 2.35 (3H, s), 1.60 (9H, s); HRMS (ESI-TOF) m/z [M + H]+: 403.1615 (Calcd for C22H22F3N2O2: 403.1633).
Methyl 4-(5-(4-Fluorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (9c)4′-Fluoroacetophenone (300 µL, 2.48 mmol) was dissolved in dry THF (6.0 mL) under ice-cold, and 60% NaH (223 mg, 5.58 mmol, 2.3 equiv.) was added. After stirring for 1 h under 0 °C, ethyl trifluoroacetate (444 µL, 3.72 mmol, 1.5 equiv.) was added, and the reaction mixture was stirred for 1 h at room temperature. The reaction mixture was acidified to pH 6 with 1 N HCl, extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was reprecipitated with EtOAc and n-hexane to afford (Z)-1,1,1-trifluoro-4-(4-fluorophenyl)-4-hydroxy-3-buten-2-one (9a) (373 mg, 1.59 mmol, 64%). 1H-NMR (400 MHz, (CD3)2CO) δ: 8.00 (2H, t, J = 7.2 Hz), 7.14 (2H, t, J = 8.8 Hz), 6.23 (1H, s).
4-(5-(4-Fluorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (9b) was synthesized from 9a (196 mg, 837 µmol) in 94% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.11 (2H, d, J = 9.2 Hz), 7.52 (2H, d, J = 8.4 Hz), 7.43 (2H, q, J = 4.8, 8.4 Hz), 7.21 (2H, t, J = 8.8 Hz), 7.05 (1H, s).
9b (316 mg, 902 µmol) was dissolved in dry methanol (MeOH), and added SOCl2 (309 µL, 4.29 mmol, 4.7 equiv.) under ice-cold. After stirring for 22 h at room temperature, the reaction mixture was quenched with sat. NaHCO3 aq., and evaporated. The residue was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc = 10 : 1) to afford 9c (196 mg, 538 µmol, 60%). Purity: 97.3%; tR: 32.1 min; 1H-NMR (400 MHz, CDCl3) δ: 8.04 (2H, d, J = 6.8 Hz), 7.38 (2H, d, J = 6.8 Hz), 7.20 (2H, t, J = 7.2 Hz), 7.05 (2H, t, J = 7.8 Hz), 6.75 (1H, s), 3.93 (3H, s); 13C-NMR (126 MHz, CDCl3) δ: 166.1, 163.3 (d, 1JCF = 251 Hz), 144.1, 144.0 (q, 2JCF = 38.4 Hz), 142.6, 130.9 (d, 3JCF = 8.4 Hz), 130.7, 130.1, 125.2, 125.1, 121.2 (q, 1JCF = 269 Hz), 116.3 (d, 2JCF = 21.7 Hz), 106.4, 52.5; 19F-NMR (373 MHz, CDCl3) δ: −62.3, −110.6; HRMS (ESI-TOF) m/z [M + H]+: 365.0915 (Calcd for C18H13F4N2O2: 365.0913).
Methyl 4-(5-(4-Chlorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (10c)(Z)-4-(4-Chlorophenyl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (10a) was synthesized from 4′-chloroacetophenone (500 µL, 3.85 mmol) in 53% yield using the method described for the preparation of 9a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.89 (2H, d, J = 8.4 Hz), 7.49 (2H, d, J = 8.8 Hz), 6.54 (1H, s).
4-(5-(4-Chlorophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (10b) was synthesized from 10a (404 mg, 1.61 mmol) in 64% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.12 (2H, d, J = 8.8 Hz), 7.53 (2H, d, J = 8.8 Hz), 7.47 (2H, d, J = 8.4 Hz), 7.40 (2H, d, J = 8.8 Hz), 7.10 (1H, s).
10c was synthesized from 10b (152 mg, 414 µmol) in 72% yield using the method described for the preparation of 9c. Purity: 95.0%; tR: 33.5 min; 1H-NMR (400 MHz, CDCl3) δ: 8.05 (2H, d, J = 9.2 Hz), 7.38 (2H, d, J = 8.8 Hz), 7.33 (2H, d, J = 8.4 Hz), 7.15 (2H, d, J = 8.4 Hz), 6.77 (1H, s), 3.93 (3H, s); 13C-NMR (126 MHz, CDCl3) δ: 166.1, 144.1 (q, 2JCF = 38.4 Hz), 143.9, 142.5, 135.8, 130.8, 130.2, 129.4, 127.4, 125.2, 125.1, 121.1 (q, 1JCF = 269 Hz), 106.5, 52.6; 19F-NMR (471 MHz, CDCl3) δ: −62.3; HRMS (ESI-TOF) m/z [M + H]+: 381.0642 (Calcd for C18H13ClF3N2O2: 381.0618).
Methyl 4-(5-(4-Bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (11c)4′-Bromoacetophenone (302 mg, 1.52 mmol) was dissolved in dry THF (4.0 mL), and 60% NaH (85 mg, 2.13 mmol, 1.4 equiv.) was added under ice-cold. After stirring for 1 h under 0 °C, ethyl trifluoroacetate (267 µL, 2.24 mmol, 1.5 equiv.) was added, and the reaction mixture was stirred for 0.5 h at room temperature. The reaction mixture was acidified to pH 6 with 1 N HCl, extracted two times with EtOAc, and washed with brine to afford (Z)-4-(4-bromophenyl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (11a) (343 mg, 1.16 mmol, 76%). 1H-NMR (400 MHz, (CD3)2CO) δ: 7.84 (2H, d, J = 8.4 Hz), 7.55 (2H, d, J = 8.8 Hz), 6.26 (1H, s).
4-(5-(4-Bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (11b) was synthesized from 11a (153 mg, 519 µmol) in 90% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.12 (2H, d, J = 9.2 Hz), 7.62 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.8 Hz), 7.33 (2H, d, J = 8.8 Hz), 7.10 (1H, s).
11c was synthesized from 11b (104 mg, 252 µmol) in 54% yield using the method described for the preparation of 9c. Purity: 96.3%; tR: 33.8 min; 1H-NMR (400 MHz, (CD3)2CO) δ: 8.09 (2H, d, J = 8.8 Hz), 7.61 (2H, d, J = 8.4 Hz), 7.54 (2H, d, J = 9.2 Hz), 7.32 (2H, d, J = 8.8 Hz), 7.10 (1H, s), 3.91 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 425.2155 (Calcd for C18H13BrF3N2O2: 425.0113).
Methyl 4-(5-Phenyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (12c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-phenyl-3-buten-2-one (12a) was synthesized from acetophenone (1.00 mL, 8.57 mmol) quantitatively using the method described for the preparation of 11a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.95 (2H, d, J = 7.2 Hz), 7.50 (1H, t, J = 7.6 Hz), 7.42 (2H, t, J = 8.0 Hz), 6.35 (1H, s).
4-(5-Phenyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (12b) was synthesized from 12a (395 mg, 1.83 mmol) in 95% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.09 (2H, d, J = 8.4 Hz), 7.51 (2H, d, J = 8.8 Hz), 7.45–7.35 (5H, m), 7.04 (1H, s).
12c was synthesized from 12b (97 mg, 292 µmol) in 52% yield using the method described for the preparation of 9c. Purity: 94.4%; tR: 32.2 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.8 Hz), 7.41–7.32 (5H, m), 7.22 (J = 6.4 Hz, 2H, d), 6.77 (1H, s), 3.92 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 347.1004 (Calcd for C18H14F3N2O2: 347.1007).
Methyl 4-(5-(4-Ethylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (13c)Sixty percent NaH (166 mg, 4.15 mmol, 2.1 equiv.) was dissolved in dry tert-butyl methyl ether (6.0 mL), and added p-ethylacetophenone (300 µL, 2.01 mmol) under ice-cold. After stirring for 1 h under 0 °C, ethyl trifluoroacetate (361 µL, 3.02 mmol, 1.5 equiv.) was added, and the reaction mixture was stirred for 2.5 h at room temperature. The reaction mixture was acidified to pH 6 with 1 N HCl, extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was reprecipitated with EtOAc and n-hexane to afford (Z)-4-(4-ethylphenyl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (13a) (193 mg, 790 µmol, 39%). 1H-NMR (400 MHz, (CD3)2CO) δ: 7.91 (2H, d, J = 8.4 Hz), 7.30 (2H, d, J = 7.6 Hz), 6.36 (1H, s), 2.68 (2H, q, J = 7.6, 15.2 Hz), 1.22 (3H, t, J = 7.6 Hz).
4-Hydrazinobenzoic acid (69 mg, 453 µmol, 1.1 equiv.) was dissolved in dry EtOH (5.0 mL), added 2 N HCl (260 µL) and 13a (99 mg, 405 µmol). After stirring for 1.5 h at 75 °C, the reaction mixture was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was dissolved in EtOAc, and n-hexane was added to remove the precipitated solid by filtration. The filtrate was evaporated to afford 4-(5-(4-ethylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (13b) (118 mg, 328 µmol, 81%). 1H-NMR (400 MHz, CD3OD) δ: 8.05 (2H, d, J = 8.8 Hz), 7.42 (2H, d, J = 8.8 Hz), 7.21 (4H, q, J = 8.8, 14.8 Hz), 6.91 (1H, s), 2.66 (2H, q, J = 7.6, 15.2 Hz), 1.22 (3H, t, J = 7.6 Hz).
13b (90 mg, 250 µmol) was dissolved in dry MeOH, and added SOCl2 (90.0 µL, 1.25 mmol, 5.0 equiv.) under ice-cold. After stirring for 23 h at room temperature, the reaction mixture was quenched with sat. NaHCO3 aq., extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc = 10 : 1) to afford 13c (57 mg, 152 µmol, 61%). Purity: 99.8%; tR: 34.0 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.8 Hz), 7.40 (2H, d, J = 8.4 Hz), 7.15 (4H, q, J = 8.0, 19.6 Hz), 6.74 (1H, s), 3.93 (3H, s), 2.66 (2H, q, J = 8.0, 15.2 Hz), 1.25 (3H, t, J = 7.6 Hz); HRMS (ESI-TOF) m/z [M + H]+: 375.1323 (Calcd for C20H18F3N2O2: 375.1320).
Methyl 4-(3-(Trifluoromethyl)-5-(4-(trifluoromethyl)phenyl)-1H-pyrazol-1-yl)benzoate (14c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(4-(trifluoromethyl)phenyl)-3-buten-2-one (14a) was synthesized from 4′-(trifluoromethyl)acetophenone (200 µL, 982 µmol) in 72% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.18 (2H, d, J = 8.0 Hz), 7.80 (2H, d, J = 8.8 Hz), 6.42 (1H, s).
4-Hydrazinobenzoic acid (112 mg, 736 µmol, 1.2 equiv.) was dissolved in dry EtOH (5.0 mL), added 2 N HCl (450 µL) and 14a (180 mg, 633 µmol). After stirring for 2 h at 75 °C, the reaction mixture was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was reprecipitated with EtOAc and n-hexane to afford 4-(3-(trifluoromethyl)-5-(4-(trifluoromethyl)phenyl)-1H-pyrazol-1-yl)benzoic acid (14b) (168 mg, 420 µmol, 66%). 1H-NMR (400 MHz, (CD3)2CO) δ: 8.12 (2H, d, J = 8.4 Hz), 7.78 (2H, d, J = 8.4 Hz), 7.62 (2H, d, J = 8.0 Hz), 7.53 (2H, d, J = 8.4 Hz), 7.20 (1H, s).
14c was synthesized from 14b (115 mg, 317 µmol) in 68% yield using the method described for the preparation of 9c. Purity: 94.4%; tR: 33.1 min; 1H-NMR (400 MHz, CDCl3) δ: 8.07 (2H, d, J = 8.4 Hz), 7.62 (2H, d, J = 8.8 Hz), 7.38 (2H, d, J = 8.8 Hz), 7.35 (2H, d, J = 8.4 Hz), 6.84 (1H, s), 3.94 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 415.0997 (Calcd for C19H13F6N2O2: 415.0881).
Methyl 4-(5-(4-Cyanophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (15c)(Z)-4-(4,4,4-Trifluoro-1-hydroxy-3-oxo-1-buten-1-yl)benzonitrile (15a) was synthesized from 4-acetylbenzonitrile (200 mg, 1.38 mmol) in 92% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.15 (2H, d, J = 8.0 Hz), 7.87 (2H, d, J = 8.4 Hz), 6.42 (1H, s).
4-(5-(4-Cyanophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (15b) was synthesized from 15a (145 mg, 601 µmol) in 61% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, CD3OD) δ: 8.09 (2H, d, J = 8.4 Hz), 7.75 (2H, d, J = 8.0 Hz), 7.48 (2H, d, J = 8.4 Hz), 7.44 (2H, d, J = 8.4 Hz), 7.12 (1H, s).
15c was synthesized from 15b (110 mg, 308 µmol) in 56% yield using the method described for the preparation of 13c. Purity: 96.9%; tR: 30.5 min; 1H-NMR (400 MHz, CDCl3) δ: 8.08 (2H, d, J = 8.4 Hz), 7.65 (2H, d, J = 8.8 Hz), 7.36 (4H, q, J = 8.8, 12.0 Hz), 6.86 (1H, s), 3.94 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 372.0980 (Calcd for C19H13F3N3O2: 372.0960).
Methyl 4-(5-(4-(Methylthio)phenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (16c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(4-(methylthio)phenyl)-3-buten-2-one (16a) was synthesized from 4′-(methylthio)acetophenone (100 mg, 602 µmol) in 30% yield using the method described for the preparation of 11a. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.07 (2H, d, J = 8.8 Hz), 7.43 (2H, d, J = 8.4 Hz), 6.88 (1H, s), 2.60 (3H, s).
4-(5-(4-(Methylthio)phenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (16b) was synthesized from 16a (36 mg, 137 µmol) in 73% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, CD3OD) δ: 8.07 (2H, d, J = 7.2 Hz), 7.43 (2H, d, J = 8.0 Hz), 7.22 (4H, q, J = 8.8, 17.6 Hz), 6.94 (1H, s), 2.48 (3H, s).
16c was synthesized from 16b (36 mg, 95.1 µmol) in 53% yield using the method described for the preparation of 9c. Purity: 95.0%; tR: 33.3 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 9.2 Hz), 7.39 (2H, d, J = 8.8 Hz), 7.17 (2H, d, J = 8.4 Hz), 7.11 (2H, d, J = 8.4 Hz), 6.73 (1H, s), 3.92 (3H, s), 2.48 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 393.0850 (Calcd for C19H16F3N2O2S: 393.0885).
Methyl 4-(5-(4-Hydroxyphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (17c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(4-hydroxyphenyl)-3-buten-2-one (17a) was synthesized from p-hydroxyacetophenone (204 mg, 1.50 mmol) in 15% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 9.34 (1H, br s), 7.95 (2H, d, J = 8.8 Hz), 6.92 (2H, d, J = 8.8 Hz).
4-Hydrazinobenzoic acid (44 mg, 289 µmol, 1.6 equiv.) was dissolved in dry EtOH (3.0 mL), added 2 N HCl (130 µL) and 17a (43 mg, 185 µmol). After stirring for 3 h at 75 °C, EtOAc was added and the precipitate was removed by filtration. The filtrate was evaporated, and the residue was reprecipitated with EtOAc and n-hexane to afford 4-(5-(4-hydroxyphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (17b) (21 mg, 60.3 µmol, 33%). 1H-NMR (400 MHz, CD3OD) δ: 8.06 (2H, d, J = 8.4 Hz), 7.42 (2H, d, J = 8.4 Hz), 7.10 (2H, d, J = 8.8 Hz), 6.84 (1H, s), 6.77 (2H, d, J = 8.4 Hz).
17c was synthesized from 17b (20 mg, 57.4 µmol) in 66% yield using the method described for the preparation of 13c. Purity: 95.9%; tR: 28.5 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.4 Hz), 7.40 (2H, d, J = 8.4 Hz), 7.09 (2H, d, J = 8.4 Hz), 6.80 (2H, d, J = 8.0 Hz), 6.71 (1H, s), 3.93 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 363.0976 (Calcd for C18H14F3N2O3: 363.0956).
Methyl 4-(5-(4-Methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (18c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(4-methoxyphenyl)-3-buten-2-one (18a) was synthesized from 4′-methoxyacetophenone (105 mg, 699 µmol) in 89% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.97 (2H, d, J = 8.8 Hz), 6.98 (2H, d, J = 9.2 Hz), 6.33 (1H, s), 3.86 (3H, s).
4-(5-(4-Methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (18b) was synthesized from 18a (127 mg, 516 µmol) in 71% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.06 (2H, d, J = 8.8 Hz), 7.43 (2H, d, J = 8.4 Hz), 7.20 (2H, d, J = 9.2 Hz), 6.92 (2H, d, J = 8.4 Hz), 6.88 (1H, s), 3.80 (3H, s).
18c was synthesized from 18b (115 mg, 317 µmol) in 68% yield using the method described for the preparation of 9c. Purity: 98.9%; tR: 31.7 min; 1H-NMR (400 MHz, CDDl3) δ: 8.03 (2H, d, J = 8.0 Hz), 7.40 (2H, d, J = 8.8 Hz), 7.14 (2H, d, J = 8.8 Hz), 6.86 (2H, d, J = 9.2 Hz), 6.71 (1H, s), 3.93 (3H, s), 3.82 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 377.1113 (Calcd for C19H16F3N2O3: 377.1113).
Methyl 4-(5-(4-Nitrophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (19c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(4-nitrophenyl)-3-buten-2-one (19a) was synthesized from 4′-nitroacetophenone (211 mg, 1.28 mmol) in 94% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.25 (2H, d, J = 8.8 Hz), 8.15 (2H, d, J = 8.4 Hz), 6.33 (1H, s).
4-(5-(4-Nitrophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (19b) was synthesized from 19a (182 mg, 697 µmol) in 90% yield using the method described for the preparation of 14b. 1H-NMR (400 MHz, CD3OD) δ: 8.24 (2H, d, J = 9.2 Hz), 8.08 (2H, d, J = 8.8 Hz), 7.55 (2H, d, J = 9.2 Hz), 7.45 (2H, d, J = 8.4 Hz), 7.16 (1H, s).
19c was synthesized from 19b (185 mg, 490 µmol) in 46% yield using the method described for the preparation of 9c. Purity: 97.8%; tR: 31.0 min; 1H-NMR (400 MHz, CDCl3) δ: 8.22 (2H, d, J = 8.8 Hz), 8.08 (2H, d, J = 8.8 Hz), 7.40 (4H, q, J = 8.8, 11.6 Hz), 6.90 (1H, s), 3.94 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 392.0880 (Calcd for C18H13F3N3O4: 392.0858).
Methyl 4-(5-(4-(Methylamino)phenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (20c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(4-(methylamino)phenyl)-3-buten-2-one (20a) was synthesized from 4′-(methylamino)acetophenone (104 mg, 697 µmol) in 91% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, CD3OD) δ: 7.84 (2H, d, J = 8.0 Hz), 6.57 (2H, d, J = 8.4 Hz), 6.27 (1H, s), 2.82 (3H, s).
4-Hydrazinobenzoic acid (73 mg, 480 µmol, 1.1 equiv.) was dissolved in dry EtOH (5.0 mL), added 2 N HCl (250 µL) and 20a (104 mg, 424 µmol). After stirring for 2.5 h at 75 °C, the reaction mixture was extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (CH2Cl2/MeOH = 10 : 1) to afford 20b (28 mg, 77.4 µmol, 18%). 1H-NMR (500 MHz, CD3OD) δ: 8.04 (2H, d, J = 8.5 Hz), 7.43 (2H, d, J = 8.5 Hz), 7.00 (2H, d, J = 8.5 Hz), 6.77 (1H, s), 6.54 (2H, d, J = 7.0 Hz), 2.76 (3H, s).
20c was synthesized from 20b (28 mg, 77.5 µmol) in 58% yield using the method described for the preparation of 9c. Purity: 98.1%; tR: 29.4 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.8 Hz), 7.43 (2H, d, J = 9.2 Hz), 7.01 (2H, d, J = 9.2 Hz), 6.65 (1H, s), 6.52 (2H, d, J = 8.8 Hz), 3.92 (3H, s), 2.85 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 376.1276 (Calcd for C19H17F3N3O2: 376.1273).
Methyl 4-(5-(4-(Dimethylamino)phenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (21c)(Z)-4-(4-(Dimethylamino)phenyl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (21a) was synthesized from p-(dimethylamino)acetophenone (198 mg, 1.21 mmol) in 72% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.84 (2H, d, J = 8.8 Hz), 6.66 (2H, d, J = 8.4 Hz, 2H), 6.24 (1H, s), 3.00 (6H, s).
4-(5-(4-(Dimethylamino)phenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (21b) was synthesized from 21a (202 mg, 779 µmol) in 85% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.06 (2H, d, J = 8.4 Hz), 7.44 (2H, d, J = 8.8 Hz), 7.08 (2H, d, J = 9.2 Hz), 6.80 (1H, s), 6.69 (2H, d, J = 9.2 Hz), 2.96 (6H, s).
21c was synthesized from 21b (198 mg, 528 µmol) in 74% yield using the method described for the preparation of 13c. Purity: 96.9%; tR: 31.4 min; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.8 Hz), 7.44 (2H, d, J = 8.8 Hz), 7.05 (2H, d, J = 9.2 Hz), 6.66 (1H, s), 6.61 (2H, d, J = 8.8 Hz), 3.93 (3H, s), 2.98 (6H, s); HRMS (ESI-TOF) m/z [M + H]+: 390.1430 (Calcd for C20H19F3N3O2: 390.1429).
Methyl 4-(5-(Cyclohex-1-en-1-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (22c)(Z)-4-(Cyclohex-1-en-1-yl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (22a) was synthesized from 1-acetyl-1-cyclohexene (100 µL, 778 µmol) in 18% yield using the method described for the preparation of 9a. 1H-NMR (400 MHz, CD3OD) δ: 6.79 (1H, br s), 5.89 (1H, br s), 2.27–2.19 (4H, m), 1.69–1.58 (4H, m).
4-(5-(Cyclohex-1-en-1-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (22b) was synthesized from 22a (25 mg, 115 µmol) in 98% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, CD3OD) δ: 8.16 (2H, d, J = 8.8 Hz), 7.63 (2H, d, J = 8.8 Hz), 6.67 (1H, s), 5.92–5.90 (1H, m), 2.16–2.08 (4H, m), 1.69–1.60 (4H, m).
22c was synthesized from 22b (37 mg, 110 µmol) in 70% yield using the method described for the preparation of 13c. Purity: 97.3%; tR: 34.3 min; 1H-NMR (400 MHz, CD3OD) δ: 8.16 (2H, d, J = 6.8 Hz), 7.65 (2H, d, J = 6.8 Hz), 6.67 (1H, s), 5.91–5.90 (1H, m), 3.95 (3H, s), 2.14–2.08 (4H, m), 1.69–1.60 (4H, m); HRMS (ESI-TOF) m/z [M + H]+: 351.1332 (Calcd for C18H18F3N2O2: 351.1320).
Methyl 4-(5-Cyclohexyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (23c)(Z)-4-Cyclohexyl-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (23a) was synthesized from cyclohexyl methyl ketone (150 µL, 1.09 mmol) in 58% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 5.52 (1H, s), 2.15–2.09 (1H, m), 1.73–1.71 (4H, m), 1.41–1.19 (6H, m).
4-(5-Cyclohexyl-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (23b) was synthesized from 23a (98 mg, 441 µmol) in 97% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.22 (1H, d, J = 8.8 Hz), 8.18 (1H, d, J = 8.8 Hz), 7.60 (2H, d, J = 8.8 Hz), 6.66 (1H, s), 2.78–2.71 (1H, m), 1.89–1.69 (4H, m), 1.57–1.24 (6H, m).
23c was synthesized from 23b (96 mg, 284 µmol) in 36% yield using the method described for the preparation of 13c. Purity: 95.8%; tR: 33.9 min; 1H-NMR (500 MHz, CDCl3) δ: 8.19 (2H, d, J = 9.0 Hz), 7.52 (2H, d, J = 8.5 Hz), 6.48 (1H, s), 3.97 (3H, s), 2.70–2.64 (1H, m), 1.86–1.70 (4H, m), 1.42–1.20 (6H, m); HRMS (ESI-TOF) m/z [M + H]+: 353.1475 (Calcd for C18H20F3N2O2: 353.1477).
Methyl 4-(5-(Pyridin-4-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (24c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(pyridin-4-yl)-3-buten-2-one (24a) was synthesized from 4-acetylpyridine (200 µL, 1.82 mmol) in 71% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.65 (2H, d, J = 5.6 Hz), 7.76 (2H, d, J = 5.6 Hz), 6.35 (1H, s).
4-Hydrazinobenzoic acid (54 mg, 355 µmol, 1.1 equiv.) was dissolved in dry EtOH (4.5 mL), added 2 N HCl (190 µL) and 24a (73 mg, 336 µmol). After stirring for 1.5 h at 75 °C, the reaction mixture was evaporated. The residue was dissolved in EtOAc and the precipitate was removed by filtration. The filtrate was evaporated to afford 4-(5-(Pyridin-4-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (24b) (69 mg, 207 µmol, 62%). 1H-NMR (400 MHz, CD3OD) δ: 8.55 (2H, d, J = 6.4 Hz), 8.12 (2H, d, J = 8.8 Hz), 7.48 (2H, d, J = 8.8 Hz), 7.34 (2H, d, J = 6.0 Hz), 7.21 (1H, s).
24c was synthesized from 24b (40 mg, 120 µmol) in 55% yield using the method described for the preparation of 13c. Purity: 95.5%; tR: 23.0 min; 1H-NMR (400 MHz, CDCl3) δ: 8.62 (2H, d, J = 5.6 Hz), 8.09 (2H, d, J = 8.8 Hz), 7.40 (2H, d, J = 8.4 Hz), 7.13 (2H, d, J = 6.0 Hz), 6.90 (1H, s), 3.95 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 348.0967 (Calcd for C17H13F3N3O2: 348.0960).
Methyl 4-(5-(1-Methyl-1H-pyrrol-3-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (25c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(1-methyl-1H-pyrrol-3-yl)-3-buten-2-one (25a) was synthesized from 3-acetyl-1-methyl pyrrole (300 µL, 2.54 mmol) in 26% yield using the method described for the preparation of 13a. 1H-NMR (500 MHz, CD3OD) δ: 7.31 (1H, br s), 6.59 (1H, s), 6.45 (1H, s), 5.97 (1H, br s), 3.66 (3H, s).
4-(5-(1-Methyl-1H-pyrrol-3-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (25b) was synthesized from 25a (49 mg, 224 µmol) in 89% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.13 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.4 Hz), 6.73 (1H, s), 6.65–6.61 (2H, m), 5.85 (1H, dd, J = 2.0, 2.8 Hz), 3.60 (3H, s).
25c was synthesized from 25b (45 mg, 134 µmol) in 81% yield using the method described for the preparation of 13c. Purity: 97.4%; tR: 30.3 min; 1H-NMR (400 MHz, CDCl3) δ: 8.09 (2H, d, J = 8.8 Hz), 7.55 (2H, d, J = 8.4 Hz), 6.60 (1H, s), 6.53–6.47 (2H, m), 5.89 (1H, dd, J = 1.6, 2.8 Hz), 3.95 (3H, s), 3.61 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 350.1131 (Calcd for C17H15F3N3O2: 350.1116).
Methyl 4-(5-(Furan-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (26c)(Z)-1,1,1-Trifluoro-4-(furan-2-yl)-4-hydroxy-3-buten-2-one (26a) was synthesized from 2-acetylfuran (200 µL, 1.99 mmol) in 51% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.64 (1H, d, J = 0.8 Hz), 7.10 (1H, d, J = 2.8 Hz), 6.54–6.53 (1H, m), 6.19 (1H, s).
4-(5-(Furan-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (26b) was synthesized from 26a (72 mg, 349 µmol) in 84% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.17 (2H, d, J = 8.0 Hz), 7.57 (1H, d, J = 2.0 Hz), 7.54 (2H, d, J = 8.4 Hz), 7.05 (1H, s), 6.49–6.48 (1H, m), 6.31 (1H, d, J = 3.6 Hz).
26c was synthesized from 26b (41 mg, 127 µmol) in 59% yield using the method described for the preparation of 13c. Purity: 96.1%; tR: 30.9 min; 1H-NMR (400 MHz, CDCl3) δ: 8.15 (2H, d, J = 8.8 Hz), 7.52 (2H, d, J = 8.8 Hz), 7.43 (1H, d, J = 1.6 Hz), 6.91 (1H, s), 6.40–6.39 (1H, m), 6.18 (J = 3.6 Hz, 1H, d), 3.96 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 337.0779 (Calcd for C16H12F3N2O3: 337.0800).
Methyl 4-(5-(Thiophen-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (27c)(Z)-1,1,1-Trifluoro-4-hydroxy-4-(thiophen-2-yl)-3-buten-2-one (27a) was synthesized from 2-acetylyhiophene (200 µL, 1.85 mmol) in 91% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.55 (1H, d, J = 3.6 Hz), 7.53 (1H, d, J = 5.2 Hz), 7.06 (1H, d, J = 4.4 Hz), 5.95 (1H, s).
4-(5-(Thiophen-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (27b) was synthesized from 27a (102 mg, 459 µmol) in 93% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.13 (2H, d, J = 8.0 Hz), 7.54–7.52 (3H, m), 7.06–7.04 (2H, m), 7.01 (1H, s).
27c was synthesized from 27b (47 mg, 139 µmol) in 59% yield using the method described for the preparation of 13c. Purity: 97.7%; tR: 31.7 min; 1H-NMR (400 MHz, CDCl3) δ: 8.10 (2H, d, J = 7.2 Hz), 7.49 (2H, d, J = 8.8 Hz), 7.38 (1H, d, J = 4.8 Hz), 6.99 (1H, t, J = 4.0 Hz), 6.88 (1H, d, J = 3.6 Hz), 6.82 (1H, s), 3.95 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 353.0571 (Calcd for C16H12F3N2O2S: 353.0572).
Methyl 4-(5-(Naphthalen-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (28c)Sixty percent NaH (237 mg, 5.93 mmol, 2.1 equiv.) and 2′-acetonaphtone (490 mg, 2.88 mmol) were dissolved in dry THF (10 mL) under ice-cold. After stirring for 1 h under 0 °C, ethyl trifluoroacetate (515 µL, 4.31 mmol, 1.5 equiv.) was added, and the reaction mixture was stirred for 29 h at room temperature. The reaction mixture was acidified to pH 6 with 1 N HCl, extracted two times with EtOAc, and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was reprecipitated with EtOAc and n-hexane, purified by silica gel flash column chromatography (n-hexane/EtOAc = 2 : 1) to afford (Z)-1,1,1-trifluoro-4-hydroxy-4-(naphthalen-2-yl)-3-buten-2-one (28a) (286 mg, 1.07 mmol, 37%). 1H-NMR (500 MHz, CD3OD) δ: 8.54 (1H, s), 8.07 (1H, d, J = 8.5 Hz), 7.99–7.88 (3H, m), 7.56–7.52 (2H, m), 6.56 (1H, s).
4-(5-(Naphthalen-2-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (28b) was synthesized from 28a (51 mg, 192 µmol) in 76% yield using the method described for the preparation of 2. 1H-NMR (400 MHz, (CD3)2CO) δ: 8.09 (2H, d, J = 8.8 Hz), 8.03 (1H, s), 7.95–7.89 (3H, m), 7.60–7.55 (4H, m), 7.37 (1H, d, J = 6.8 Hz), 7.16 (1H, s).
28c was synthesized from 28b (38 mg, 99.4 µmol) in 79% yield using the method described for the preparation of 13c. Purity: 97.9%; tR: 34.4 min; 1H-NMR (400 MHz, CDCl3) δ: 8.01 (2H, d, J = 6.8 Hz), 7.85–7.77 (4H, m), 7.55–7.51 (2H, m), 7.43 (2H, d, J = 7.2 Hz), 7.19 (1H, d, J = 8.8 Hz), 6.87 (1H, s), 3.91 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 397.1163 (Calcd for C22H16F3N2O2: 397.1164).
Methyl 4-(5-(2,3-Dihydro-1H-inden-5-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (29c)(Z)-4-(2,3-Dihydro-1H-inden-5-yl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (29a) was synthesized from 5-acetylindane (150 µL, 999 µmol) in 29% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, CD3OD) δ: 7.78–7.72 (2H, br), 7.23 (1H, d, J = 7.2 Hz), 6.32 (1H, br s), 2.94–2.91 (4H, m), 2.13–2.06 (2H, m).
4-(5-(2,3-Dihydro-1H-inden-5-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (29b) was synthesized from 29a (50 mg, 195 µmol) in 96% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.04 (2H, d, J = 9.2 Hz), 7.42 (2H, d, J = 8.8 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.15 (1H, s), 6.99 (1H, d, J = 8.0 Hz), 6.88 (1H, s), 2.93–2.84 (4H, m), 2.11–2.04 (2H, m).
29c was synthesized from 29b (44 mg, 118 µmol) in 42% yield using the method described for the preparation of 13c. Purity: 98.3%; tR: 35.1 min; 1H-NMR (500 MHz, CDCl3) δ: 8.03 (2H, d, J = 8.5 Hz), 7.41 (2H, d, J = 8.5 Hz), 7.16 (1H, d, J = 7.5 Hz), 7.11 (1H, s), 6.93 (1H, d, J = 7.5 Hz), 6.72 (1H, s), 3.92 (3H, s), 2.93–2.85 (4H, m), 2.12–2.06 (2H, m); HRMS (ESI-TOF) m/z [M + H]+: 387.1334 (Calcd for C21H18F3N2O2: 387.1320).
Methyl 4-(5-(Benzo[d][1,3]dioxol-5-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoate (30c)(Z)-4-(Benzo[d][1,3]dioxol-5-yl)-1,1,1-trifluoro-4-hydroxy-3-buten-2-one (30a) was synthesized from 3′,4′-(methylenedioxy)acetophenone (199 mg, 1.21 mmol) in 80% yield using the method described for the preparation of 13a. 1H-NMR (400 MHz, (CD3)2CO) δ: 7.55–7.35 (2H, m), 6.83 (2H, d, J = 8.4 Hz), 6.22 (1H, s), 6.02 (2H, s).
4-(5-(Benzo[d][1,3]dioxol-5-yl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic acid (30b) was synthesized from 30a (103 mg, 396 µmol) in 91% yield using the method described for the preparation of 13b. 1H-NMR (400 MHz, CD3OD) δ: 8.07 (2H, d, J = 8.0 Hz), 7.44 (2H, d, J = 8.4 Hz), 6.88 (1H, s), 6.84–6.77 (2H, m), 6.74 (1H, s), 5.99 (2H, s).
30c was synthesized from 30b (101 mg, 268 µmol) in 82% yield using the method described for the preparation of 13c. Purity: 99.5%; tR: 31.4 min; 1H-NMR (400 MHz, CDCl3) δ: 8.05 (2H, d, J = 8.4 Hz), 7.41 (2H, d, J = 8.8 Hz), 6.79–6.64 (4H, m), 6.00 (2H, s), 3.93 (3H, s); HRMS (ESI-TOF) m/z [M + H]+: 391.0894 (Calcd for C19H14F3N2O4: 391.0906).
Cell CultureHT-1080 human sarcoma cells (purchased from American Type Culture Collection, Manassas, VA, U.S.A.) were cultured in Dulbecco’s modified Eagle’s medium (Wako Pure Chemical Corporation, Osaka, Japan). The culture medium was supplemented with 10% fetal bovine serum, and the cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C.
In Vitro WST-8 AssayIn vitro cytotoxicity was examined using a colorimetric assay with the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan), according to the manufacturer’s instructions. In the WST-8 assay, triplicate wells were used for each concentration, and all assays were performed at three times. Briefly, HT-1080 cells were seeded at a density of 4 × 103 cells/well in a 96-well plate and cultured for 24 h. Serial dilutions of each compound, dissolved in dimethyl sulfoxide, were added to the culture medium at concentrations of 3.125–100 µM. After 48 h of incubation, the medium was replaced with fresh medium containing the WST-8 reagent. After 1 h, the absorbance in each well was measured at 460 nm using a microplate spectrophotometer, ImmunoMini NJ-2300 (BioTec, Tokyo, Japan).
The percentage of cell growth inhibition was calculated by applying the following formula: percentage of cell growth inhibition = (1 − [T/C]) × 100, where C and T are the mean absorbance values of the control and treated groups, respectively. The IC50 value was measured graphically from the dose-response curve with at least three drug concentration points.
Gelatin ZymographyHT-1080 cells (1 × 105 cells/well) were seeded in a 6-well tissue culture plate and cultured in 10% fetal bovine serum/Dulbecco’s modified Eagle medium (FBS/DMEM) overnight. After washing twice with phosphate-buffered saline (PBS), the cells were serum-starved with 0.5% FBS/DMEM for 6 h, and then treated with the inhibitors (25 µM) for 24 h. The conditioned medium (CM) was analyzed by gelatin zymography, as previously described.19) MMP-2 and MMP-9 levels in CM were measured by adding an equal volume of sample buffer. The samples were separated by electrophoresis on an sodium dodecyl sulfate (SDS)-polyacrylamide gel containing gelatin (Difco, Sparks, MD, U.S.A.) labeled with Alexa Fluor 680 (Molecular Probes). The gels were processed and monitored using an Odyssey infrared imaging system (LI-COR, Lincoln, NE, U.S.A.) against the normalized density of the target band in the control sample. These assays were performed at three times.
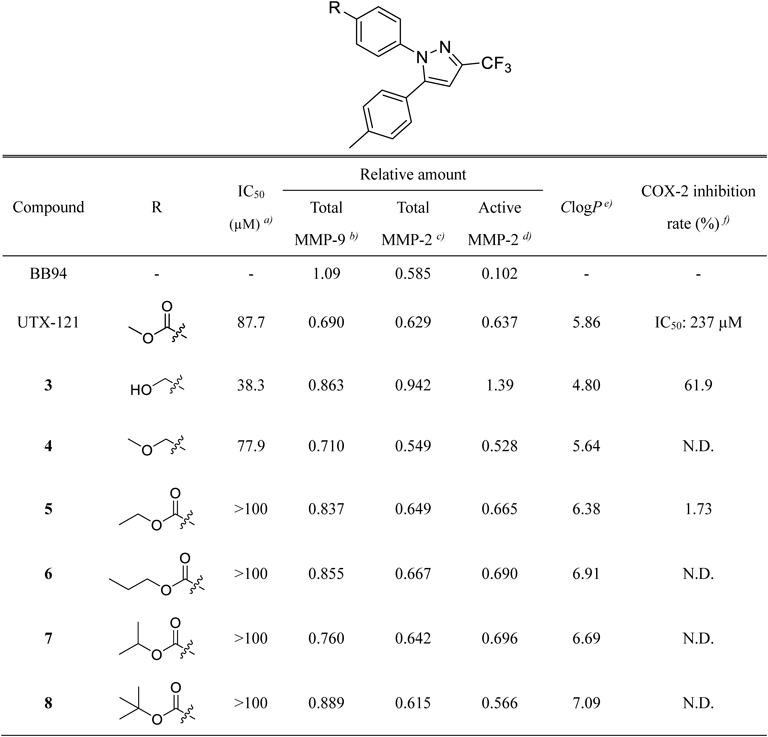
Batimastat (BB94) acts as a direct inhibitor of various MMPs such as MMP-1, MMP-2, MMP-9, MMP-7, and MMP-3. BB94 can bind the zinc ion in the active site of MMPs.22) To evaluate the inhibitory activity of the derivatives upon MMP-2/9 production, BB94 was used as the positive control.
COX-2 Inhibition AssayCOX-2 inhibitory activity was assessed using the COX-2 (human) Inhibitor Screening Assay Kit (Cayman Chemical, Ann Arbor, MI, U.S.A.) according to the manufacturer’s instructions. In brief, COX-2 was inactivated by placing it in boiling water for 3 min. Compounds (10 µL; final concentration: 200 µM) dissolved in DMSO were added to each test tube, and 10 µL of DMSO was added to the control and background tubes. All tubes were incubated for 10 min at 37 °C in a heat block. Arachidonic acid (10 µL) was added to the reaction tubes to initiate the reaction. The tubes were quickly mixed and incubated for 2 min at 37 °C. Saturated stannous chloride solution (30 µL) was added to each reaction tube to stop enzyme catalysis. All tubes were removed from the heat block, vortexed, and incubated for 5 min at room temperature. The amount of prostaglandin in the tubes was evaluated by enzyme-linked immunosorbent assay (ELISA).19) The COX-2 inhibitory assay for each compound was carried out in triplicate.