Reviews
Imbuing an Old Heterocycle with the Power of Modern Catalysis: An Isoxazolidin-5-one Story
2021 Volume 69 Issue 12 Pages 1160-1169
Details
2021 Volume 69 Issue 12 Pages 1160-1169
Isoxazolidin-5-ones have been regarded as β-amino acid surrogates owing to their labile N–O bond. While many efforts have been devoted to the catalytic enantioselective synthesis of the core of this heterocycle, its further transformation has been less explored, especially in the context of catalysis. This review summarizes the author’s research on the development of catalytic reactions using isoxazolidin-5-ones as substrates. Asymmetric catalysis has proven effective for C–C bond formation at the carbonyl α-carbon. Catalytic asymmetric allylation and direct Mannich-type reactions have been developed. Further, the resulting products have been readily converted into the corresponding quaternary β2,2-amino acids. Moreover, isoxazolidin-5-ones have been identified as alkyl nitrene precursors in the presence of a suitable metal catalyst. The generated metallonitrene undergoes either the electrophilic amination of the aromatic ring or aliphatic C–H insertion, affording a series of cyclic β-amino acids. A remarkable difference in chemoselectivity between rhodium and copper alkyl nitrenes has also been demonstrated, highlighting the unique nature of the underexplored reactive intermediates. The various linear and cyclic β-amino acids obtained through the study are likely to find great utility in a broad range of chemical sciences.
Over the past decades, the evolution of catalysis has revolutionized the way chemists synthesize molecules. Catalysts have provided a new mode of activation that enables the transformation of otherwise inert compounds.1) They have also been sophisticated in orchestrating a highly ordered transition state to realize various selectivities including enantioselectivity.2) Given the incredible feats accomplished, it is unsurprising that three Nobel prizes have been awarded for catalysis-related research in the 21st century.
Isoxazolidin-5-ones are isoxazol-derived, five-membered heterocycles,3,4) whose preparation was documented as early as in 1909.5) Since their characteristic N–O bond is readily cleaved under reductive conditions, isoxazolidin-5-ones have been considered as β-amino acid equivalents upon ring opening.6) The prevalence of β-amino acids and their derivatives in biologically active compounds7) such as β-lactams has led to the development of several cyclization approaches for the synthesis of functionalized isoxazolidin-5-ones. In some studies, asymmetric syntheses relying on either chiral auxiliary8–13) or asymmetric catalysis14–16) have been achieved. The catalytic asymmetric synthesis of isoxazolidin-5-ones has been summarized in a recent review.17)
In contrast to the catalytic construction of the five-membered ring, the use of the heterocycle itself as a substrate in catalytic reactions has been less explored until recently. Significant progress has been made in the last five years along this line,18–24) making it possible to synthesize structurally diversified β-amino acids otherwise difficult or impossible to obtain. This review focuses on this emerging approach, highlighting the author’s work in recent years. This review is divided into two main sections. The first elucidates the catalytic asymmetric α-functionalization of the aforenoted heterocycle. In this case, the resulting adducts are converted to the corresponding linear β-amino acids by taking advantage of known reductive conditions. The other section deals with redox-neutral cyclization for the synthesis of cyclic β-amino acids. This is achieved by a combination of the reductive cleavage of the N–O bond and the oxidation of a C–H bond. Both reaction paradigms have further established the utility of isoxazolidin-5-ones as β-amino acid surrogates. Notably, the effectiveness of sophisticated catalysts plays a major role in eliciting the hidden reactivity of the heterocycle and controlling the resulting reactive intermediate.
β-Amino acids are structurally classified according to the number and position of their substituents.25) Among them, β2,2-amino acids bearing two geminal substituents at the α-carbon are the most difficult to attain, and the available catalytic asymmetric synthesis route remains limited in terms of structural diversity.26) In addition, most existing reports do not directly pertain to peptide synthesis,27–29) which is unfortunate because peptides are the most sought-after products of β-amino acids.30)
At the outset of the author’s research program, the α-quaternization of racemic isoxazolidin-5-ones was envisioned as a viable approach to tackle these existing problems (Chart 1). A literature precedent regarding stoichiometric, chiral-auxiliary-based alkylation has set a solid foundation for the α-quaternization.31) The resulting geminally substituted chiral heterocycle would be readily converted to the corresponding quaternary β2,2-amino acids upon the N–O bond cleavage.6) The successful enolization of the heterocycle and subsequent stereoselective α-functionalization encouraged the implementation of its catalytic enantioselective variant from racemic isoxazolidin-5-ones.

To investigate the feasibility of catalytic asymmetric C–C bond formation at the α-position of isoxazolidin-5-ones, decarboxylative asymmetric allylation was targeted.32–36) Conditions screening revealed that a chiral palladium complex ligated with a Trost ligand37) promoted the desired allylation in a highly stereoselective manner38) (Fig. 1). The developed catalytic conditions exhibited a broad scope with respect to the α-substituents. A series of substituted benzyl groups was accommodated at the α-position, and the allylated products were obtained in good yields and enantioselectivities. Heteroaryl, alkenyl, and alkyl groups were also tolerated.

An obvious benefit associated with the allyl group is its diversified downstream transformations that allow for the synthesis of various linear β2,2-amino acids (Chart 2). Combined with the reductive cleavage of the N–O bond, β-amino acids with varied chain lengths and oxidation states were readily obtained.

Another unique asset linked with the N–O bond is amide-bond forming ligation with α-ketoacids. The α-ketoacid-hydroxylamine ligation39) allowed for direct access to 9-fluorenylmethyloxycarbonyl (Fmoc)-protected α-β dipeptide 12. The purity of its stereochemistry was readily enriched by crystallization. The obtained dipeptide was amenable to a standard Fmoc-based solid-phase peptide synthesis (Fmoc-SPPS), providing a hybrid α/β peptide in good overall yield (Chart 3). This protocol represents the first catalytic asymmetric synthesis of β2,2-amino acid suitable for Fmoc-SPPS.

The decarboxylative allylation described in the preceding subsection necessitates the prefunctionalization of the substrates before the catalytic asymmetric transformation. Obviating the activation step would lead to a more atom-economical and straightforward approach.40) In addition, more diversified products would be obtained just by changing reaction partners, ideally with a slight modification of catalytic conditions. Hence, this direct catalytic reaction was investigated (Chart 4).

From the outset, the diastereo- and enantioselective construction of contiguous stereocenters was targeted, and the direct Mannich-type reaction of α-aryl pronucleophile 14 and isatin-derived ketimine 15 was investigated. Extensive catalyst screening identified that metal-based catalytic systems failed to produce the Mannich adduct with acceptable enantioselectivity, albeit the reactivity was excellent. In contrast, chiral organocatalysts afforded the desired product in high selectivity. The dimeric catalyst, (DHQD)2PYR, was found to be optimal in terms of enantioselectivity. The scope of the developed conditions was broad with respect to the substituents at both the α-position and the imine41) (Fig. 2). A broad range of ketimines was tolerated, giving the corresponding products in good selectivity and almost perfect stereoselectivity. In addition, various substituted aryls, including a heteroaryl, can be introduced at the α-position.
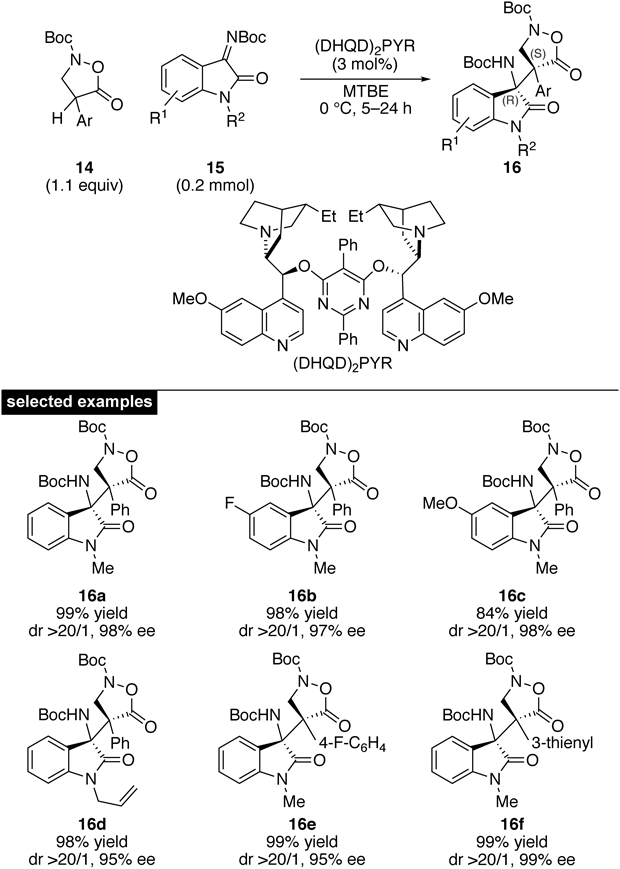
A slight modification of the catalytic conditions also allowed for enantioselective conjugate addition to nitrostyrene 18, demonstrating the generality of the catalytic system (Chart 5).

One of the drawbacks of the organocatalytic Mannich-type reaction is the requirement for an α-aryl group, which enhances the acidity of the α-proton and thus enables facile enolization with a rather weak Brønsted base. To expand the pronucleophile scope with respect to α-substituents, further catalyst screening was performed using an α-benzyl type substrate. A combination of a chiral silver complex and lithium phenoxide has emerged as a promising catalytic system. Specifically, the best catalytic system comprised two ligands: a Taniaphos ligand for the silver and bis(phosphine oxide) for the lithium base. Imines derived from a series of aromatic aldehydes underwent smooth and enantioselective direct Mannich-type reactions, delivering the corresponding products in high yields and enantioselectivities42) (Fig. 3). Regarding the α-substituent, various benzyl-type substituents afforded the products in good enantioselectivities, but the use of a substrate with a homobenzyl group resulted in diminished enantioselectivity, presumably due to the unfavorable orientation toward an attractive π-interaction important for the high enantioselectivity. Another disadvantage of the current protocol lies in its modest diastereoselectivity.

The Mannich product obtained by this reaction contains two Boc-protected nitrogen atoms. The unique nature of the N–O bond allowed for the differential transformation of the two amines (Chart 6).

The previous section describes the catalytic asymmetric synthesis of 4,4-disubstituted isoxazolidine-5-ones. Given the labile nature of the N–O bond under reductive conditions, the synthesis is equivalent to the synthesis of the corresponding linear β2,2-amino acids (Chart 7a). The ring opening is reliable, but the requirement for an external reductant does not make the most of the oxidation state of hydroxylamine. Hence, a redox-neutral transformation based on a combination of the reductive cleavage of the N–O bond and the oxidation of a C–H bond in the same molecule was envisioned. It would produce the corresponding cyclic β-amino acid in a redox-neutral manner (Chart 7b). The resulting product is also a saturated N-heterocycle having a carboxylic acid at the β-carbon. In consideration of the importance of saturated N-heterocycles in medicinal chemistry,43) even racemic synthesis is expected to find broad utility.44,45)

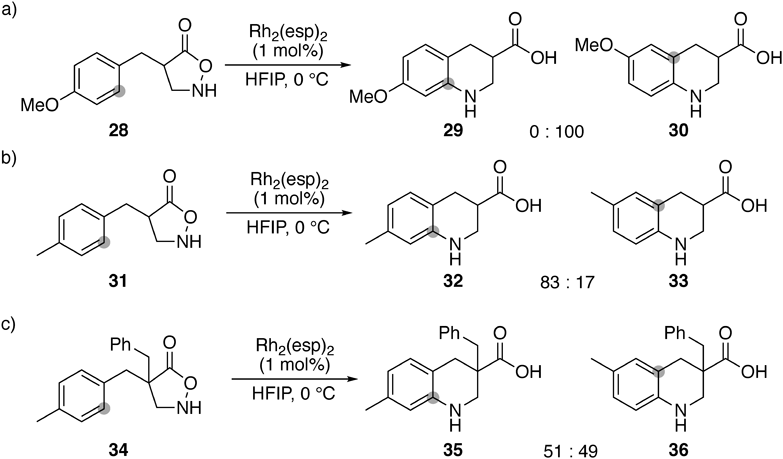
Extensive conditions screening revealed that catalytic amounts of Rh2(esp)246) promoted the electrophilic amination of 4-benzyl isoxazolidin-5-one in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP)47) (Fig. 4). The rhodium conditions were sensitive to electronics on the aromatic ring, and substrates having an electron-withdrawing group generally exhibited lower reactivity. Nevertheless, the corresponding cyclic compound was obtained even from a strongly electron-deficient nitro-substituted arene. The observed reactivity tendency pointed out the involvement of electrophilic nitrogen species as a reactive intermediate.48) A structurally unique heteroaromatic embedded bicyclic compound was also obtained. Furthermore, the introduction of an α-substituent at the carbonyl group did not cause any problem for the cyclization.

The cyclization mechanism was originally considered to be the formation of a direct six-membered ring from the ortho-carbon. The formation of unexpected skeletal rearranged product 30 from 28, however, requested a modification of the proposed mechanism (Chart 8a). Furthermore, the ratio of the rearranged product differed depending on the substrate structure (Charts 8b, c).
The skeletal rearranged product was proposed to have formed via a spirocyclic intermediate that was generated from an ipso-attack to a provisional rhodium nitrene49) (Chart 9). The ipso-cyclization mechanism was supported by Hammett analysis using differentially substituted dibenzyl substrates.50) In this mechanism, the product distribution is determined by the difference in the rate of C–N and C–C bond migration to the adjacent carbon.

Other experimental evidence for the ipso-cyclization was obtained from attempted five- and seven-membered ring formations (Chart 10). The formation of five-membered ring 38 was not observed, presumably due to the unfavorable formation of four-membered spiro intermediate 39. In contrast, seven-membered ring 41 was obtained together with N-Ph pyrrolidine 42. The latter compound likely formed from the spiro intermediate, followed by aromatization to generate a tertiary cation, which is trapped by nitrogen. Thus, the formation of pyrrolidine 42 strongly supports the ipso-attack mechanism, rather than the ortho-attack mechanism.

The two-step mechanisms depicted in Chart 9 suggest the possibility of isolating the spiro intermediate if it is sufficiently stabilized. When 4-OH substituted substrates 43 were used, spirocyclic β-amino acids 44 were obtained as isomerized dienone forms (Fig. 5). A series of substituted phenols was converted into the dearomatized products in good yields. For the unsymmetrically substituted substrate, the diastereoselectivity remained low. The products contained both a nucleophilic amine and a highly electrophilic enone, and the key for the success of the isolation was the formation of a zwitterion between carboxylic acid and amine, preventing oligomerization.51)

Spirocyclic scaffolds have garnered increasing attention in medicinal chemistry.52) Notably, spirocyclic β-amino acids 44 obtained from phenol substrates 43 are structurally unique and should serve as useful building blocks. If the electrophilic nitrogen species described in the preceding subsection had indeed been a rhodium nitrene, intramolecular C(sp3)–H insertion would be a viable approach toward broader spirocyclic β-amino acids (Chart 11a). Although Rh-catalyzed C(sp3)–H insertion has been well established,53,54) the structures of nitrene precursors are largely limited to carbamates and sulfamates (Chart 11b). Alkyl nitrenes having C–H bond(s) next to the nitrogen are known to be unstable55) (Chart 11c), and their use in organic synthesis has been underexplored.56–62)

When suitably substituted isoxazolidin-5-ones were subjected to a catalytic amount of Rh2(esp)2 in HFIP, the expected spirocyclic β-prolines were exclusively formed63) (Fig. 6). Carbocycles of various sizes were introduced into the spiro junction. Nitrogen-, oxygen-, and sulfur-containing heterocycles can also be merged with a β-proline skeleton. Furthermore, the presence of an α-substituent did not affect the reaction efficiency.

The rhodium conditions were also suitable for the synthesis of spirocyclic β-homoprolines if the substrates were changed to 3-substituted isoxazolidin-5-ones (Fig. 7).

Two experimental observations revealed an important reactivity profile for the rhodium-catalyzed C–H insertion. First, rhodium nitrene prefers forming a five-membered ring even when more electronically activated, methine C–H bond is available for the six-membered ring formation (Chart 12a). Second, electrophilic amination is faster than C–H insertion, and a mixture of products is obtained if the substrate has two reaction sites (Chart 12b).

The lack of chemoselectivity under the rhodium conditions shown in Chart 12 led to the development of new catalytic conditions.64) Given that different metals often exhibit distinct reactivity profiles, catalysts comprising metals other than rhodium were screened. While many metal salts lack sufficient catalytic activity, a copper salt has emerged as a promising catalyst. After extensive optimization studies, the optimal conditions were identified to employ a pyridine-benzoxazole hybrid ligand in HFIP65) (Fig. 8). The new copper conditions were found to selectively promote aromatic electrophilic amination, even in the presence of potentially reactive functionalities, including benzylic and tertiary C(sp3)–H bonds and olefins. In the case of propargylic compound 27m, further cyclization took place to afford spiro lactone 53. Similar to the rhodium conditions, the copper conditions are also sensitive to the electronic nature of the aromatic ring, suggesting the involvement of an electrophilic nitrogen species as a reactive intermediate.

Drastic changes in chemoselectivity are often realized by changes in the reactive intermediates. Given the literature precedents regarding iron-catalyzed radical aromatic amination using a hydroxylamine reagent,66,67) the radical-based mechanism was substantially feasible. In this mechanism, copper salt acts as an electron shuttle to promote the homolytic cleavage of the N–O bond and aromatization of the intermediate (Chart 13a). Three experimental observations disfavored the naked nitrogen-centered radical mechanism (Charts 13b–d). First, the addition of butylated hydroxytoluene (BHT), a radical scavenger, did not suppress the reactivity. Second, the use of N-Me substrate 56 instead of N-H one 54 did not lead to any reactivity under otherwise identical conditions. Finally, catalytic asymmetric desymmetrization was possible when a chiral bis(oxazoline) (BOX) ligand was used for the copper.68) These results collectively disallow the involvement of a naked, nitrogen-centered radical.

Despite the less preferable results for the radical-based mechanism, no experimental support for the copper nitrene mechanism was obtained.69) To gain insights into the reaction intermediate, a substrate having a phenyl group next to the nitrogen was synthesized (Chart 14). The designed substrate was expected to undergo facile isomerization to an imine if the corresponding alkyl nitrene was generated under the reaction conditions. When the 13C-labeled compound was subjected to copper conditions, a clean conversion to labeled acetophenone was observed. The lack of decarboxylation with benzoylacetic acid under the copper conditions indicated that the ketone was formed by the generation of an alkyl nitrene, which isomerizes to an imine, followed by decarboxylation and hydrolysis. The observation of the ketone supports the formation of a copper-bound alkyl nitrene under the reaction conditions.

Regarding the cyclization mechanism with an interim copper nitrene, no rearranged products were observed during the substrate scope study. Moreover, 4-OH substituted substrate 60 provided a mixture of spirocyclic and six-membered ring products (Chart 15a). These results, which contradict those obtained in the case of rhodium, strongly suggest that a mechanism different from that of the rhodium reactions underlies the copper-catalyzed cyclization. A similarly distinct chemoselectivity was observed with a homologated substrate. The rhodium conditions exclusively afforded pyrrolidine 64 via C(sp3)–H insertion, whereas the copper conditions only formed spirocyclic dienone 65 (Chart 15b).

The above results supported the involvement of copper-bound alkyl nitrene, but did not reveal the pathways of ortho- or ipso-cyclization for the C–N bond formation. Computational studies together with kinetic experiments showed that dicopper nitrene is involved in the catalytic cycle.70) They also revealed a Curtin–Hammett type energy landscape wherein both cyclization pathways are energetically accessible, but the ipso intermediate is not productive because of the high activation energy required for the subsequent C–N bond migration. Therefore, according to the computations, all products are formed through the ortho-cyclization, which is consistent with the lack of rearranged product formation (Chart 16). Although the same molecule was obtained from the same substrate and a similar metallonitrene, a mechanistic study revealed that different cyclization pathways were entailed depending on the nature of the metallonitrenes.

The chemistry of isoxazolidin-5-ones with stoichiometric amounts of reagents were considerably advanced in the 1980s when the synthesis of β-lactam antibiotics was extensively investigated. However, the further transformation of this century-old heterocycle has not undergone significant progress in spite of substantial developments in catalysis. This review has summarized recent developments in catalysis with respect to isoxazolidin-5-ones based on the author’s work. Asymmetric catalysts, both metal-based catalysts and organocatalysts, are successful at forming a new C–C bond at the α-position of the carbonyl group in a highly enantioselective manner, allowing for the synthesis of linear β2,2-amino acids upon reductive N–O bond cleavage. Furthermore, isoxazolidin-5-ones have been identified as alkyl nitrene precursors in the presence of suitable metal catalysts. The resulting reactive intermediate undergoes either electrophilic amination or C–H insertion. Redox-neutral cyclization proves useful for the synthesis of cyclic β-amino acids. The chemoselectivity of metallonitrenes differs remarkably depending on the metal center. Many β-amino acids described in this review are structurally unique and otherwise difficult to synthesize. Given the broad application of amino acids, they are likely to find specific utility in a wide range of disciplines in chemical science.
This review of the author’s work was written by the author upon receiving the 2021 Pharmaceutical Society of Japan Award for Young Scientists.
I would like to express my sincere gratitude to Prof. Dr. Masakatsu Shibasaki (Institute of Microbial Chemistry, Tokyo), who offered generous support, constant encouragement, and constructive suggestions essential to achieving the results described in this review. I am extremely grateful to all my coworkers, whose names appear in the reference section, for their dedicated efforts in this research. This work was financially supported by JSPS KAKENHI Grant Numbers JP16K18856, JP18K14878, and JP20K06957.
The author declares no conflict of interest.