Notes
Preparation of Isodehydrodigallic Acid Using Ullmann Condensation
2021 Volume 69 Issue 3 Pages 298-301
Details
2021 Volume 69 Issue 3 Pages 298-301
Isodehydrodigallic acid, which is an important component of several ellagitannin compounds, was easily synthesized using a classical Ullmann condensation reaction.
Several types of polyphenolic compound have been isolated from higher plants, and their chemical structures have been elucidated using NMR techniques.1) Among them, ellagitannin compounds can be isolated from the aqueous phase of plant extracts. Ellagitannins display remarkable structural complexity, involving glucose moieties and gallic acid (1) derivatives. Furthermore, ellagitannins exhibit diverse and valuable biological properties, such as antioxidant,2–4) antitumor,5–7) antimicrobial,8) and antiviral9,10) activities. The yields of ellagitannins are low when they are extracted from natural sources; methods for chemical synthesis of ellagitannins and related compounds should therefore be developed.
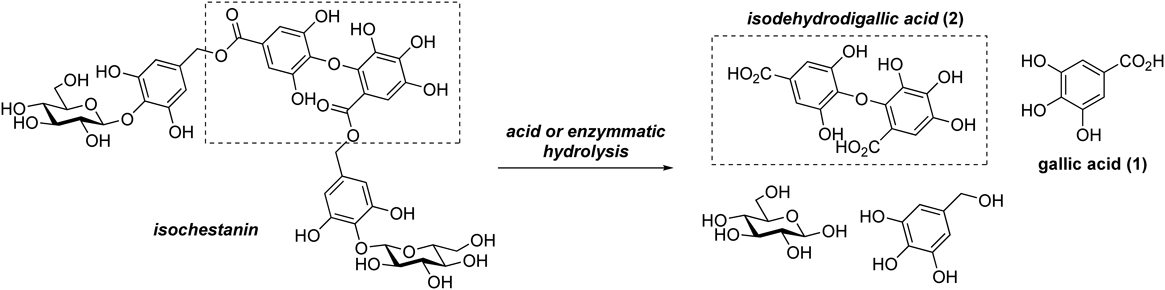
The structures of several ellagitannin compounds contain either C–C or C–O–C bonds connecting the aromatic rings of two gallic acid moieties. Hexahydroxydiphenoyl (HHDP) and dehydrodigalloyl (DHDG) groups are commonly found in ellagitannin molecules. Isodehydrodigalloyl (isoDHDG),11–13) which has a tetra-ortho-substituted diaryl ether structure, is also found in naturally occurring ellagitannins. An example of a natural product containing the isoDHDG group is isochestanin, which was previously isolated from Castanea mollissima (Feng et al.). Isodehydrodigallic acid was identified as a hydrolyzation product of isochestanin14) (Chart 1).
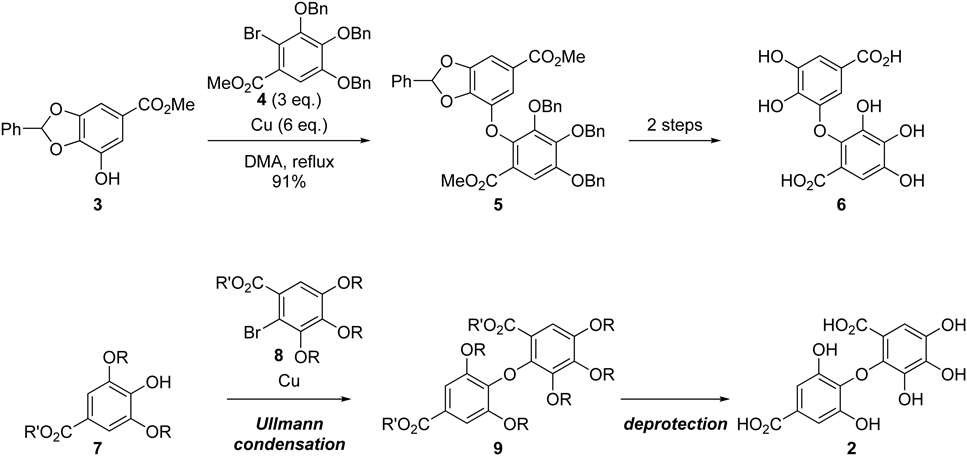
To synthesize tetra-ortho-substituted diaryl ether, a process for synthetic C–O–C bond formation between two aromatic rings that is capable of overcoming steric hindrance must be established. Several reports have presented methods for preparing sterically hindered diaryl ether.15–17) Synthesis of diaryl ether derived from gallic acid using Ullmann condensation was reported.18) However, the desired diaryl ether was only obtained in 33% yield in this method. On the other hand, we explored for the condition of classical Ullmann condensation19) and established a simple method for diaryl ether synthesis, in which diaryl ether (5) was synthesized from phenol (3) and bromide (4) using copper powder. Moreover, after a two-step conversion process, dehydrodigallic acid (6) was formed20) (Chart 2). In the present study, we applied a similar method to synthesize isodehydrodigallic acid (2) which possesses sterically more hindered property. Steric hindrance had to be overcome at the key reactive positions; thus, this article describes the preparation of the phenol (7) and bromide (8) compound precursors, as well as the key Ullmann condensation reaction for the synthesis of isodehydrodigallic acid (2) (Chart 2).
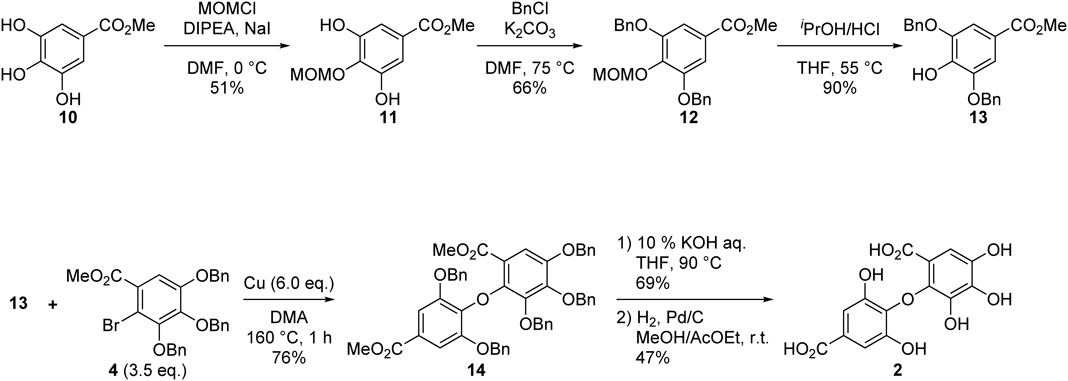
We first generated the phenol compound required to synthesize the desired diaryl ether. A phenolic hydroxyl group at the 4-position of methyl gallate (10) was protected with a methoxymethyl (MOM) group to obtain compound 11. Then, the remaining phenolic hydroxy groups at the 3- and 5-positions of 11 were protected with benzyl groups to form 12, followed by deprotection of the MOM group with aqueous HCl to produce 13. We then synthesized the coupling partner, bromide 4, from methyl gallate (10) via a two-step process by the reported procedure.20) Following the preparation of compounds 13 and 4, we conducted the Ullmann condensation reaction under the following conditions: copper (6.0 equivalent (eq.)) and N,N-dimethylacetamide (DMA) at 160 °C for 1 h. We obtained a 76% yield of the desired ether compound 14, as well as 52% of debrominated compound 4 and a small amount of dimerized compound 4 as a by-product. Finally, a two-step process involving saponification and hydrogenolysis concluded the synthesis of isodehydrodigallic acid (2) (Chart 3).
In this study, we chemically synthesized isodehydrodigallic acid using the Ullmann condensation reaction, and demonstrated its effectiveness for synthesizing sterically hindered diphenyl ethers.
The melting points of each compound were measured using a Yanagimoto micro-melting point hot-plate. IR spectra were recorded using a Shimadzu FTIR-8400 spectrophotometer. NMR spectra were obtained using a JEOL α-400 (400 MHz) instrument. Chemical shifts were expressed in δ parts per million (ppm), with tetramethylsilane (TMS) as an internal standard. Elemental analyses were performed using an Elementar vario MICRO cube. The electron impact (EI)MS analysis was performed using the JMS-AX505HAD instrument (JEOL, Tokyo, Japan). Silica gel column chromatography was carried out using Wakogel® C-200 or 60N as the stationary phase. TLC analysis was performed on Silicagel 70 F254 (Wako, Osaka, Japan) plates. Solvents were dried using a standard procedure.
Methyl 3,5-Dihydroxy-4-methoxymethylbenzoate (11)A solution containing compound 10 (5.00 g, 27.2 mmol), N,N-dimethylformamide (DMF) (100 mL), diisopropylethylamine (DIPEA) (5.68 mL, 27.2 mmol) and NaI (4.07 g, 27.2 mmol) was stirred for 30 min at 0 °C. MOMCl (2.06 mL, 32.6 mmol) was then added to the solution and stirred for 10 min at the same temperature. The mixture was neutralized by addition of sat. aq NH4Cl and the resulting mixture was then extracted using ethyl acetate (AcOEt). The organic layer was washed with brine, dried over MgSO4, and then evaporated to yield a crude residue. The residue was recrystallized from AcOEt to generate a colorless solid of 11 (3.03 g, 51%). mp 126.0–129.0 °C (AcOEt). IR (KBr): υmax 3300, 2955, 1904, 1678, 1281, 773 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 7.21 (1H, s, Ph), 6.76 (1H, s, Ph), 5.14 (2H, s, CH2), 3.88 (3H, s, CH3), 3.72 (3H, s, CH3). 13C-NMR (100 MHz, CDCl3) δ: 166.5, 148.2, 137.3, 127.0, 109.8, 99.2, 57.6, 52.2. Anal. Calcd for C10H12O6: C, 52.64; H, 5.30. Found: C, 52.67; H, 5.27.
Methyl 3,5-Dibenzyloxy-4-methoxymethylbenzoate (12)A solution containing compound 11 (1.4 g, 6.05 mmol), K2CO3 (3.0 g, 21.8 mmol) and DMF (14 mL) was stirred for 30 min at room temperature. benzyl chloride (1.6 mL, 13.9 mmol) was added to the solution and stirred for 4 h at room temperature. The mixture was acidified by addition of 10% HCl (aq), and then poured into water and extracted using AcOEt. The organic layer was washed with brine, dried over MgSO4, and then evaporated to yield a crude residue. The residue was recrystallized from AcOEt to generate a colorless solid of 12 (1.6 g, 66%). mp 104.3–105.2 °C (AcOEt). IR (KBr) υmax 2951, 2886, 1716, 1261, 757 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 7.46–7.30 (12H, m, Ph), 5.20 (2H, s, CH2), 5.14 (4H, s, PhCH2 × 2), 3.89 (3H, s, CH3), 3.47 (3H, s, CH3). 13C-NMR (100 MHz, CDCl3) δ: 166.5, 152.3, 139.8, 136.4, 128.5, 128.1, 127.6, 125.5, 108.6, 98.3, 71.1, 57.2, 52.2. Anal. Calcd for C24H24O6: C, 70.58; H, 5.92. Found: C, 70.45; H, 5.97.
Methyl 3,5-Dibenzyloxy-4-hydroxybenzoate (13)A solution containing 12 (1.5 g, 3.72 mmol) and tetrahydrofuran (THF) (11 mL) was stirred for 30 min at room temperature. A mixture of iPrOH and conc. HCl (v/v = 50/1, 33 mL) was added to the solution, which was stirred for 1.5 h at 55 °C. The mixture was cooled to room temperature and NaHCO3 (aq) was added. The mixture was then poured into water and extracted using AcOEt. The organic layer was washed with brine, dried over MgSO4, and evaporated to yield a crude residue. The residue was recrystallized from AcOEt to generate a colorless solid of 13 (1.2 g, 90%). mp 133.2–134.5 °C (AcOEt). IR (KBr): υmax 3438.8, 2954.7, 2874.7, 1699.2, 746.4 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 7.46–7.33 (12H, m, Ph), 5.94 (1H, s, OH), 5.18 (4H, s, PhCH2 × 2), 3.87 (3H, s, CH3). 13C-NMR (100 MHz, CDCl3) δ: 166.7, 145.9, 140.2, 136.2, 128.6, 128.3, 127.8, 120.9, 108.7, 71.4, 52.1. Anal. Calcd for C22H20O5: C, 72.52; H, 5.53. Found: C, 72.42; H, 5.50.
Methyl 3,4,5-Tri(benzyloxy)-2-(2,6-(benzyloxy)-4-(methoxycarbonyl)phenoxy) Benzoate (14)The flask was charged with 13 (100 mg, 0.274 mmol), 4 (512 mg, 0.960 mmol), Cu (105 mg, 1.65 mmol), and DMA (0.7 mL). The resulting mixture was stirred under N2 at 160 °C for 1 h, and then cooled to room temperature, filtered, and washed with AcOEt. The mixture was acidified by adding 10% HCl (aq), and then poured into water and extracted with AcOEt. The organic layer was washed with brine and dried over MgSO4. A residue formed following evaporation, which was purified using silica gel column chromatography (CH2Cl2/hexane/EtOAc = 1 : 1 : 0.25) to generate a yellow oil of 14 (170 mg, 76%). IR (neat): υmax 3028, 2950, 1716, 1590, 1434, 1336, 1213, 1113, 747, 703 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 7.45–7.12 (28H, m, ArH), 4.99 (2H, s, CH2), 4.92 (4H, br, CH2), 4.86 (2H, s, CH2), 4.83 (2H, s, CH2), 3.90 (3H, s, CH3), 3.70 (3H, s, CH3). 13C-NMR (100 MHz, CDCl3) δ: 166.6, 165.9, 149.4, 147.9, 146.7, 144.8, 141.1, 137.2, 137.1, 136.6, 136.4, 128.5, 128.2, 128.0, 127.5, 127.4, 117.6, 110.2, 109.1, 75.3, 75.1, 71.2, 71.1, 52.1, 52.0. High resolution (HR)MS-EI (m/z): Calcd for C51H44O10 [M]+ 816.2934; Found, 816.2929.
2-(2,6-Benzyloxy-4-carbaxyphenoxy)-3,4,5-tribenzyloxybenzoic Acid (15)A solution containing compound 14 (166 mg, 0.203 mmol) and THF (15 mL) was stirred for 30 min at room temperature. Then, 10 mL of 10% KOH (aq) was added to the solution and stirred for 48 h at room temperature. NaHCO3 (aq) was then added to the mixture, which was poured into water and extracted with AcOEt. The organic layer was washed with brine, dried over MgSO4, and evaporated to give a residue which was recrystallized from CH2Cl2. A white solid of 15 (110 mg, 69%) was generated. mp 185.0–187.5 °C (CH2Cl2). IR (KBr): υmax 1684, 1433, 1113, 733, 696 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 7.45–6.85 (28H, m, ArH), 5.06 (2H, s, CH2), 4.98 (2H, s, CH2), 4.92 (2H, br, CH2), 4.77 (2H, br, CH2), 4.67 (2H, s, CH2). 13C-NMR (100 MHz, CDCl3) δ: 171.1, 166.9, 150.1, 148.5, 146.8, 146.2, 143.9, 140.2, 136.8, 136.6, 136.3, 135.6, 128.6, 128.5, 128.4, 127.8, 127.7, 116.1, 110.6, 108.8, 75.4, 74.9, 71.1, 70.9. Anal. Calcd for C49H40O10: C, 74.61; H, 5.11. Found: C, 74.58; H, 5.08.
Isodehydrodigallic Acid (2)A solution containing compound 15 (400 mg, 0.507 mmol) and methanol (MeOH)/AcOEt (1 : 1, 16 mL) was stirred at room temperature. Then, 10% Pd/C (40 mg) was added to the solution and stirred for 1.5 h at room temperature. The reaction mixture was filtered and washed with MeOH/AcOEt, and the organic layer was then evaporated to give a residue which was recrystallized from H2O, resulting in a colorless solid of 2 (81 mg, 47%). mp 199.0–200.0 °C (H2O). IR (KBr): υmax 3246, 2525, 1690, 1607, 1348, 1262, 729 cm−1. 1H-NMR (400 MHz, acetone-d6 + D2O) δ: 7.10 (2H, s, ArH), 6.97 (1H, s, ArH). 13C-NMR (100 MHz, acetone-d6 + D2O)21) δ: 170.6, 167.7, 150.6, 142.3, 139.9, 139.6, 139.2, 138.6, 127.5, 114.5, 110.5, 108.2. Anal. Calcd for C14H10O10: C, 49.72; H, 3.19. Found: C, 49.55; H, 2.93.
A part of this study was financially supported by THE HOKURIKU BANK Grant-in-Aid for Young Scientists for H. A.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.