Notes
Direct N1-Selective Alkylation of Hydantoins Using Potassium Bases
2021 Volume 69 Issue 4 Pages 407-410
Details
2021 Volume 69 Issue 4 Pages 407-410
Hydantoins, including the antiepileptic drug phenytoin, contain an amide nitrogen and an imide nitrogen, both of which can be alkylated. However, due to the higher acidity of its proton, N3 can be more easily alkylated than N1 under basic conditions. In this study, we explored methods for direct N1-selective methylation of phenytoin and found that conditions using potassium bases [potassium tert-butoxide (tBuOK) and potassium hexamethyldisilazide (KHMDS)] in tetrahydrofuran (THF) gave N1-monomethylated phenytoin in good yield. The applicable scope of this reaction system was found to include various hydantoins and alkyl halides. To explore the function of methylated hydantoins, the effects of a series of methylated phenytoins on P-glycoprotein were examined, but none of methylated products showed inhibitory activity toward rhodamine 123 efflux by P-glycoprotein.
Hydantoins (1) are a major scaffold with various applications1,2) (Fig. 1). The structure is found in pharmaceuticals and pesticides, and modifications such as alkylation, arylation, and halogenation have been made at the N1, N3, and 5 positions to obtain desired activities. Phenytoin (1a) is a 5,5-diphenyl analog of 1 and is used as an anticonvulsant drug for the treatment of epilepsy. As frequently seen in antiepileptic drugs, 1a was reported as a weak substrate of P-glycoprotein (P-gp), which acts as a drug efflux pump.3,4) According to the X-ray structure of P-gp and docking studies,5,6) hydrophobic interactions and hydrogen bonding are important between P-gp and phenytoin and related compounds. Therefore, alkylation of one or both of the N1 and N3 positions of these compounds is expected to affect their pharmacokinetic behavior.
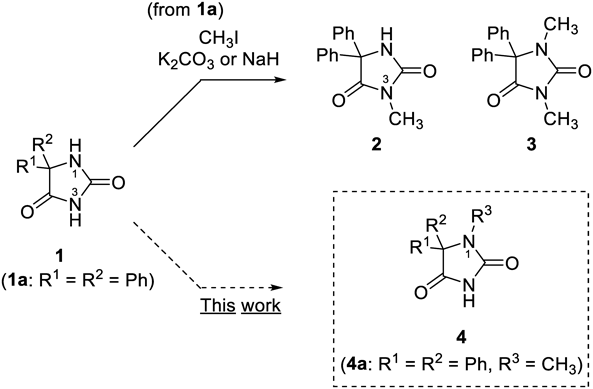
In conventional alkylation using alkyl halides under basic condition, N3 can be more easily alkylated than N1 because of the more acidic proton on N3. For example, treatment of 1a with CH3I in the presence of a weak base such as K2CO3 gives N3-monomethylated product 2, and a mixture of 2 and dimethylated 3 is obtained when stronger bases such as NaH are used.7,8) For the selective preparation of mono-N1-alkylated hydantoins such as 4, including N1-monomethylated phenytoin (4a), two main method have been utilized: 1) hydantoin-ring formation using N-substituted substrates9–13); 2) N1-alkylation of N3-protected hydantoins by aminals or tert-alkyl groups followed by deprotection of N3.14–18) To date, examples of N1-selective alkylation of unprotected hydantoins are limited. Crosignani et al.19) reported the synthesis of N1-methylated hydantoin as an intermediate of various N3-alkyl-N1-methylhydantoin derivatives.
Positron emission tomography (PET) experiments using 11C-labeled 1a showed increased brain uptake in rat after inhibition of P-gp by tariquidar.20) PET tracers that are weak P-gp substrates are useful tools for assessing P-gp function based on overexpression. A hydrogen bond between Tyr307 and the N3-hydrogen of phenytoin (1a) was estimated by in silico screening.21) Thus, N1-selective alkylation of hydantoins would enable their modification while leaving N3 unsubstituted, thus allowing for effective hydrogen bonding to P-gp. Furthermore, the development of new methodology for rapid methylation reactions would be useful for preparing compounds labeled with short-lived isotopes such as 11C. In this note, we report our studies on N1-selective alkylation of hydantoins 1, primarily phenytoin (1a), using various bases and alkyl halides in order to realize a simple method for the selective and rapid preparation of N1-alkylated phenytoin (Fig. 1), and investigate the inhibitory activity of methylated phenytoins toward P-gp using rhodamine 123.
We began our investigation by using the reaction system reported by Crosignani et al., where N-unsubstituted hydantoin was selectively methylated at N1 using lithium bis(trimethylsilyl)amide (LiHMDS) as base in tetrahydrofuran (THF) in 47% yield (room temperature (r.t.), 6 h).19) Phenytoin (1a) was treated with LiHMDS (2.2 equivalent (equiv)) and CH3I (1.2 equiv) in THF at r.t. for 6 h to give a mixture of 1a, N1-methyl 4a, and 1,3-dimethylphenytoin (3) in a ratio of 12 : 83 : 5 (Table 1, run 1). The ratio was estimated from integration in the 1H-NMR spectra of the crude products. After purification, 4a was isolated in 66% yield. The structure of 4a were confirmed through two dimensional (2D)-NMR heteronuclear multiple bond connectivity (HMBC) experiments (see Supplementary Materials) and comparison with the reported melting point.13) In this reaction, generation of the mono-N3-methylated product 2 was not observed. Next reaction conditions in shorter reaction period for short-lived isotope was employed (5 min) with a reduced amount of CH3I (0.94 equiv) to suppress dimethylation, and desired 4a was obtained as the sole product with lower conversion (1a : 4a = 60 : 40) (run 2). Use of other disilazide bases (XHMDS: X = Na, K) improved the yield of 4a, with KHMDS giving the best results among them. tert-Butoxide bases (tBuOX: X = Li, Na, K) were tested as milder bases, and N1 selectivity and higher yield were estimated when using tBuOK, where the reactivity seems parallel to the ion pairing constants for the lithium, sodium, and potassium tert-butoxide (in dimethyl sulfoxide (DMSO))22) (runs 5–7). When 2.0 equiv of tBuOK and 1.2 equiv of CH3I was used, desired 4a was obtained in 79% isolated yield (run 8). Based on these results, tBuOK was selected as the base for further examination.
 | |||
|---|---|---|---|
| Run | Base | Ratio (1a : 4a : 3)b) | Isolated 4a (%)c) |
| 1d) | LiHMDS | 12 : 83 : 5 | 66 |
| 2 | LiHMDS | 60 : 40 : 0 | — |
| 3 | NaHMDS | 52 : 48 : 0 | — |
| 4 | KHMDS | 34 : 66 : 0 | 69 |
| 5 | tBuOLi | 84 : 16 : 0 | — |
| 6e) | tBuONa | 45 : 55 : 0 | — |
| 7 | tBuOK | 23 : 76 : 0 | 73 |
| 8f) | tBuOK | 5 : 93 : 2 | 79 |
a) Compound 1a, base solution (2.0 equiv), and CH3I (0.94 equiv) were used unless otherwise noted. b) Ratio was estimated from integration in the 1H-NMR spectra of the crude products. c) Yield based on 1a. d) Reactions were carried out using LHMDS (2.2 equiv) and CH3I (1.2 equiv) for 6 h. e) Solid tBuONa was used. f) 1.2 equiv of CH3I was used.
We next examined the effects of solvents and additives (Table 2). In toluene, the reaction gave 4a in low yield, probably due to the low solubility of 1a (run 2). When tBuOH was used as a protic solvent, the reaction was also sluggish (run 3). On the other hand, undesired N3-methylated 2 was obtained when N,N-dimethylformamide (DMF) was used (run 4). The reaction in the presence of 18-crown-6 in THF did not afford desired 4a, and 1,3-dimethylated 3 (28%) and N3-methylated 2 (44%) were obtained (run 5).
 | ||||
|---|---|---|---|---|
| Run | Solvent | Additive | Ratio (1a : 4a : 2 : 3)b) | Isolated 4a (%)c) |
| 1c) | THF | None | 23 : 76 : 0 : 0 | 73 |
| 2 | Toluene | None | 84 : 16 : 0 : 0 | 14 |
| 3 | tBuOH | None | 87 : 11 : 2 : 0 | 10 |
| 4 | DMF | None | 16 : 0 : 84 : 0 | — |
| 5d) | THF | 18-Crown-6 | 8 : 0 : 45 : 47 | — |
a) Compound 1a, tBuOK (2.0 equiv), additive (2.0 equiv), and CH3I (0.94 equiv) were used. b) Ratio was estimated from integration in the 1H-NMR spectra of the crude products. c) Table 1, run 7. d) Isolated yield: 2 (44%), 3 (28%).
We next examined reactions using other substrates with 1.2 equiv of alkyl halides (Table 3). Reaction of 5-methyl-5-phenylhydantoin (1b) gave N1-methylated 4b as the sole product in lower yield (27%) with recovering 1b (56%) (run 1). It could be because N1–H of 1a, which could be activated by two phenyl groups at C5, would be more acidic than that of 1b with one phenyl group. The reactions of phenytoin (1a) using other alkyl halides such as benzyl bromide, allyl bromide, and bromoacetate were rather slow. The reaction with benzyl bromide gave the desired N1-benzylated 4c was obtained as the major product (53%) with a small amount of dibenzylated 5 (4%) in 6 h (run 2). Lower reactivity but N1-selectivity were observed when allyl bromide was used, and N1-allylated 4d was obtained in 32% yield with recovering 1a (21%) (run 3). Generation of other by-products were not observed in the crude products in both cases. On the other hand, the desired N1-alkylated product 4e was not obtained when using ethyl bromoacetate, and instead N3-alkylated 6 (55%) and dialkylated 7 (5%) were obtained (run 4). The reactivity of methyl, benzyl, and allyl halides were reported as methyl ≈ allyl < benzyl.23,24) The lower reactivity of benzyl and allyl halides in this case would be because of steric effect between alkyl groups in alkyl halides and two phenyl groups in 1a.
 | ||||
|---|---|---|---|---|
| Run | Hydantoin | R3-X | Time | Isolated yield (%) |
| 1 | 1b | CH3I | 5 min | 4b (27) |
| 2 | 1a | PhCH2Br | 6 h | 4c (53), 5 (4) |
| 3 | 1a | CH2=CHCH2Br | 6 h | 4d (32) |
| 4 | 1a | BrCH2CO2Et | 2 h | 6 (55), 7 (5) |
a) Hydantoin, tBuOK (2.0 equiv), and R3-X (1.2 equiv) were used.
The origin of the N1-selectivity of this reaction is not clear, but the use of THF as solvent appeared to be a factor in the selectivity. As deduced from the acidity of hydantoins, N3–H will be more easily deprotonated by tBuOK than N1–H to generate potassium salt 8. X-ray crystallographic analysis of phenytoin sodium (the corresponding sodium salt of 8)25) showed the formation of a N3–Na bond, and intramolecular coordination of the oxygen in C(4)=O to Na was also observed. A similar coordinated structure might be formed in the reaction of 1a and tBuOK in THF, which is a less polar solvent than DMF, and the reaction at N3 would then be hampered by the formation of the coordinated structure. The second equivalent of base would deprotonate N1–H, and the more reactive N1 anion in 9 could react with CH3I to give N1-methylated 4a as the major product (Fig. 2). Addition of 18-crown-6 would assist the dissociation of the N3–K bond, leading to the generation of a more naked anion on N3 to give N3-methylated 2 and 1,3-dimethylated 3, as in when using DMF, which is a more polar solvent than THF (Table 2, runs 4 and 5). The reaction using bromoacetate, which has an ester group as the site of coordination to the potassium ion, also gave similar N3 selectivity (Table 3, run 4).

We examined the inhibitory activity of 1a and methylated products 2, 3, and 4a toward P-gp. We used LLC-PK1 and LLC-GA5-COL150 cells as low and high P-gp-expressing cells, respectively.26,27) Rhodamine 123 (7) and verapamil were used as a substrate and inhibitor of P-gp, respectively, and the fluorescent activity of 7 was measured in the presence of phenytoins. High uptake of 7 was observed in all the LLC-PK1 cells. In contrast, low uptake of 7 was observed in the LLC-GA5-COL150 cells in the presence of a series of methylated phenytoins. High uptake of 7 was observed even in the LLC-GA5-COL150 cells in the presence of verapamil. These results suggest these phenytoin (1a) and the derivatives have no inhibitory activity toward rhodamine 123 efflux by P-gp.
In conclusion, we optimized the reaction conditions for N1-selective alkylation on hydantoins such as phenytoin (1a) and found that reaction systems using potassium bases (tBuOK, KHMDS) in THF gave N1-methylated 4a in good yield. The methylation reaction of phenytoin (1a) proceeded rapidly, and the reaction using benzyl and allyl bromides gave the corresponding N1-alkylated product after longer reaction times. Methylated phenytoins were examined for inhibitory activity against P-gp, but none of the methylated products inhibited rhodamine 123 efflux by P-gp. Even though no inhibitory activity of phenytoin (1a) and the methylated derivatives 2, 3 and 4a toward P-gp efflux of rhodamine 123 (7) was not observed in this experiment, it is under discussion whether phenytoin (1a) can be a substrate for P-gp, and there are several reports to support it.3,20) Thus, it is necessary to confirm whether a series of phenytoin derivatives 2, 3, and 4a using PET conditions, especially for 4a with more acidic N3–H. N3-Methylated 2 and dimethylated 3 can be prepared in conventional method, and the present rapid and selective methylation to the corresponding N1-methylated derivative 4a will provide an effective method for preparing short-lived 11C-labeled hydantoins with a more acidic hydrogen which could form more stable hydrogen bond.
To a solution of phenytoin (1a, 100 mg, 0.40 mmol) in THF (2.0 mL), tBuOK (Aldrich, 1 M solution in THF, 0.80 mL, 0.80 mmol) was added at r.t. After 3 min, CH3I (30 µL, 0.48 mmol) in THF (0.1 mL) was added at r.t. and the mixture was stirred at r.t. for 5 min. One molar HCl (2 mL) was added and the whole was extracted with AcOEt (2 × 10, 1 × 5 mL). The combined organic layer was washed with brine (1 × 5 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and the crude product was purified by column chromatography (n-hexane–AcOEt 2 : 1) to give 4a as colorless solids (83 mg, 79%).
1-Methyl-5,5-diphenylimidazolidine-2,4-dione (4a)Colorless solids. mp 216.5–218.0 °C (lit13) 223–224 °C). IR (KBr) ν (cm−1) 3024, 1768, 1703. 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.77 (3H, s, CH3), 7.26–7.31 (4H, m, Ar–H), 7.38–7.42 (6H, m, Ar–H), 9.31 (1H, s, NH). 13C-NMR (125 MHz, CDCl3) δ (ppm) 26.4, 75.8, 128.2, 128.8, 128.9, 136.1, 155.9, 174.1. High resolution electrospray ionization (HRESI)MS m/z 267.1130 (Calcd for C16H14N2O2: 267.1134).
3-Methyl-5,5-diphenylimidazolidine-2,4-dione (2)Pale yellow solids. mp 218.5–219.0 °C (lit8) 223–224 °C). IR (KBr) ν (cm−1) 3287, 1773, 1700. 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.10 (3H, s, CH3), 6.11 (1H, s, NH), 7.32–7.38 (10H, m, Ar–H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 25.0, 70.3, 126.8, 128.5, 128.8, 139.0, 156.9, 173.4.
1,3-Dimethyl-5,5-diphenylimidazolidine-2,4-dione (3)Yellow solids. mp 190.5–191.5 °C (lit8) 196–198 °C). IR (KBr) ν (cm−1) 1867, 1711. 1H-NMR (500 MHz, CDCl3) δ (ppm) 2.80 (3H, s, CH3), 3.14 (3H, s, CH3), 7.24–7.26 (4H, m, Ar–H), 7.37-7.41 (6H, m, Ar–H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 25.3, 26.7, 74.6, 128.2, 128.6, 128.7, 128.8, 136.4, 156.3, 173.7.
1,5-Dimethyl-5-phenylimidazoline-2,4-dione (4b)Colorless solids, mp 185–186.5 °C (lit28) 186–188 °C). IR (KBr) 3209, 1773, 1719. 1H-NMR (500 MHz, CDCl3) δ (ppm) 1.85 (3H, s, CH3), 2.81 (3H, s, CH3), 7.30–7.55 (5H, m, Ar–H), 8.02 (1H, s, NH). 13C-NMR (125 MHz, CDCl3) δ (ppm) 20.2, 25.3, 68.2, 126.1, 129.0, 12.9.3, 136.2, 156.2, 175.7. HRESIMS m/z 205.0969 (Calcd for C11H13N2O2: 205.0977).
1-Benzyl-5,5-phenylimidazoline-2,4-dione (4c)Yellow solids. mp 217–218 °C (lit13) 212–214 °C). IR (KBr) ν (cm−1) 3177, 1773, 1707. 1H-NMR (500 MHz, CDCl3) δ (ppm) 4.54 (2H, s, CH2), 6.77 (2H, d, J = 7.3 Hz, Ar–H), 6.99–7.06 (3H, m, Ar–H), 7.25–7.33 (10H, m, Ar–H), 9.21 (1H, s, NH). 13C-NMR (125 MHz, CDCl3) δ (ppm) 45.2, 76.9, 126.9, 127.8, 127.9, 128.6, 128.7, 128.9, 136.3, 136.4, 156.3, 174.1. HRESIMS m/z 343.1443 (Calcd for C22H19N2O2: 343.1447).
1,3-Dibenzyl-5,5-phenylimidazoline-2,4-dione (5)A colorless oil. IR (neat) ν (cm−1) 1765, 1719. 1H-NMR (500 MHz, CDCl3) δ (ppm) 4.54 (2H, s, CH2), 4.77 (2H, s, CH2), 6.77 (2H, d, J = 6.9 Hz, Ar–H), 6.95–7.06 (3H, m, Ar–H), 7.17-7.19 (4H, m, Ar–H), 7.25–7.39 (11H, m, Ar–H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 43.0, 45.4, 75.5, 126.9, 127.8, 127.9, 128.0, 128.2, 128.4, 128.6, 128.6, 128.8, 136.1, 136.5, 136.7, 156.3, 173.5. HRESIMS m/z 433.1911 (Calcd for C29H25N2O2, 433.1916).
1-Allyl-5,5-diphenylimidazoline-2,4-dione (4d)Colorless solids. mp 187.5–188.0 °C. IR (KBr) 3277, 1773, 1719. 1H-NMR (500 MHz, CDCl3) δ (ppm) 3.93–3.94 (2H, m, –CH2–), 4.77–4.83 (2H, m, =CH2), 5.10–5.18 (1H, m, =CH), 7.30–7.41 (10H, m, Ar–H), 7.83 (1H, s, N–H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 44.1, 76.2, 117.6, 128.4, 128.7, 129.0, 131.6, 136.7, 155.8, 174.2. HRESIMS m/z 293.1285 (Calcd for C18H17N2O2: 293.1290).
Ethyl 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)acetate (6)Yellow solids. mp 184.5–185.5 °C (lit29) 185–186 °C). IR (KBr) 3180, 1773, 1752, 1718. 1H-NMR (500 MHz, CDCl3) δ (ppm) 1.25 (3H, t, J = 7.1 Hz, CH3), 4.22 (2H, q, J = 7.1 Hz, CH2), 4.29 (2H, s, CH2), 6.63 (1H, s, NH), 7.34–7.43 (10H, m, Ar–H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 14.2, 40.0, 62.2, 70.9, 127.3, 128.9, 190.0, 138.9, 155.7, 167.0, 173.3. HRESIMS m/z 339.1340 (Calcd for C19H19N2O4: 339.1345).
Diethyl 2,2′-(2,4-Dioxo-5,5-diphenylimidazolidine-1,3-diyl)diacetate (7)A colorless oil. IR (neat) 1784, 1752, 1727. 1H-NMR (500 MHz, CDCl3) δ (ppm) 1.04 (3H, t, J = 7.1 Hz, CH3), 1.27 (3H, t, J = 7.1 Hz, CH3), 3.80 (2H, q, J = 7.1 Hz, CH2), 4.08 (2H, s, CH2), 4.23 (2H, q, J = 7.1 Hz, CH2), 4.36 (2H, s, CH2), 7.34–7.46 (10H, m, Ar–H). 13C-NMR (125 MHz, CDCl3) δ (ppm) 13.8, 14.0, 40.2, 42.8, 61.3, 62.0, 75.2, 128.6, 128.9, 129.1, 136.3, 155.2, 166.7, 167.1, 172.9. HRESIMS m/z 425.1708 (Calcd for C23H25N2O6: 425.1713).
This work was partially supported by JSPS KAKENHI Grant Number JP 19K08139. We thank Ms. Tomoko Amimoto, the Natural Science Center for Basic Research and Development (N-BARD), Hiroshima University for the measurement of HRESIMS.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials (NMR charts, P-gp assay).