Experimental
BiologyEvaluation of mAChR Positive Allosteric Modulator ActivityPAM activity of the test substances for the human and rat mAChRs were evaluated as described previously.26) Briefly, Ca2+ signal transduction induced by CCh in human and rat mAChRs receptor-expressing CHO-K1 cells in the absence or presence of the test substance was monitored using the FLIPR™ system. PAM activity was determined by dividing the EC50 value of CCh in the absence of the test substance by the EC50 value in the presence of the test substance. For example, when the EC50 value of CCh in the absence of the test substance is 0.2 µM and the EC50 value of CCh in the presence of the test substance is 0.01 µM, the PAM activity is indicated by a 20-fold shift.
Effects on Transmural Electrical Field Stimulation-Induced Contraction of Isolated Rat Bladder TissueAll experimental procedures performed on the animals were approved by the Institutional Animal Care and Use Committee of Astellas Pharma Inc., which has been awarded Accreditation Status by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. All efforts were made to minimize animal numbers and suffering.
Evaluation of the pharmacological effects of 3g on EFS-induced contraction of isolated rat bladder tissue was conducted as described previously.26) Briefly, a strip of bladder tissue isolated from a Sprague-Dawley (SD) female rat was suspended in an organ bath filled with 10 mL of Krebs–Henseleit solution and bubbled with 95% O2 and 5% CO2, and maintained at 37 °C. After stabilizing at an initial tension of 1 g, contraction was induced twice with 60 mM KCl. The strip was then washed and stabilized, and contraction was induced with EFS at 20 V (stimulation frequency of 8 Hz, pulse width of 0.3 ms, and stimulation time of 10 s). The voltage was adjusted to induce a contractile amplitude that was about 50% of the contractile response at 20 V. After stabilization, 3g (final concentration 3, 10, and 30 µM) was added. Each subsequent concentration of the test substance was cumulatively administered after the contractile response obtained at the lower concentration had stabilized. The effect of 3g was evaluated using the average of the EFS-induced contractions before and after application.
Pharmacokinetics StudyPharmacokinetics characterization of compound 3g was conducted in female SD rats (SD SPF rats Crl: CD(SD), 8 weeks of age). Compound 3g was intravenously and orally administered at 1 mg/kg in a mixture of dimethyl sulfoxide (DMSO)/Cremophor/saline (10/10/10). The animals were fasted prior to administration. After each administration, blood was drawn at the assigned time points up to 24 h, and the concentration of compound 3g in plasma was determined using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. Pharmacokinetics parameters were calculated from the plasma concentrations for each animal by noncompartmental analysis using the pharmacokinetics software Phoenix WinNonlin.
Conformational Energy CalculationThe four possible planar conformations of compounds 2 and 3a as shown in Figs. 2 and 4 respectively were prepared by MOE.32) Each conformation was optimized with the HF/6-31G** model chemistry and the potential energy of the optimized structure was calculated at the MP2/6-31++G** level of theory. All calculations were performed with the Gaussian 09 program package33) and the data were analyzed with the Gaussview 5.0 molecular visualization program.34) The potential energy difference between two conformers was calculated to identify the most stable conformer.
Chemistry1H-NMR spectra were recorded on a JEOL JNM-EX400 spectrometer and were referenced against an internal standard, tetramethylsilane. The abbreviations used for the signal patterns are as follows: s, singlet; br, broad; d, doublet; t, triplet; q, quartet; dd, double doublet; m, multiplet. Mass spectra were recorded on a JEOL LX-2000 mass spectrometer. Elemental analyses were performed using a Yanako MT-5 microanalyzer (C, H, N) and a Yokogawa IC-7000S ion chromatographic analyzer (halogens). Where analyses are indicated by symbols, the analytical results were within ±0.4% of the theoretical values. Electrospray ionization (ESI) positive high-resolution (HR)MS were obtained using a Waters LCT Premier. Column chromatography was performed using Wakogel C-200, Merck silica gel 60, or Fuji Silysia ODS-DM1020T. All reactions were performed using commercially available reagents and solvents without further purification.
4-[3-Fluoro-5-(trifluoromethyl)phenyl]-1,3-thiazol-2-amine (5)To a mixture of 1-[3-fluoro-5-(trifluoromethyl)phenyl]ethanone (4, 78 g) and tetrahydrofuran (625 mL) was added phenyltrimethylammonium tribromide (143 g), followed by stirring at room temperature for 1 h. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The obtained compound and ethanol (625 mL) were mixed, and thiourea (35 g) was added thereto, followed by stirring at 65 to 75 °C for 2 h. The reaction mixture was cooled on ice, and water (625 mL) was added. A 1 M sodium hydroxide (600 mL) solution was added, and the mixture was stirred for 30 min. The solid was collected by filtration, and ethanol (30% aqueous, 600 mL) was added to dissolve the solid at 76 °C. The obtained solution was cooled to room temperature and stirred overnight. The mixture was cooled on ice and stirred for 2 h, and the precipitated solid was collected by filtration to obtain 5 (56.9 g, 57%) as a white solid: 1H-NMR (CDCl3) δ: 5.00 (2H, s), 6.85 (1H, s), 7.20–7.26 (1H, m), 7.64–7.70 (1H, m), 7.82–7.86 (1H, m); ESI-MS m/z 263 (M + H)+.
N-{4-[3-Fluoro-5-(trifluoromethyl)phenyl]-1,3-thiazol-2-yl}acetamide (6)4-[3-Fluoro-5-(trifluoromethyl)phenyl]-1,3-thiazol-2-amine (5, 2.8 g), pyridine (10 mL), and acetic anhydride (4 mL) were mixed, followed by stirring at 60 °C for 1 h. The reaction mixture was cooled to room temperature, water was added, and the resulting solid was collected by filtration. The solid was washed with methanol and collected by filtration to obtain 6 (2.9 g, 88%) as a white solid: 1H-NMR (DMSO-d6) δ: 2.18 (3H, s), 7.62 (1H, d, J = 8.6 Hz), 7.98 (1H, s), 8.04 (1H, d, J = 10.0 Hz), 8.11 (1H, s), 12.32 (1H, s); ESI-MS m/z 305 (M + H)+.
N-(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)acetamide (7)A mixture of N-{4-[3-fluoro-5-(trifluoromethyl)phenyl]-1,3-thiazol-2-yl}acetamide (6, 2.8 g), acetic acid (20 mL), a 36% aqueous formaldehyde solution (3.6 mL), and acetic anhydride (4.4 mL) was stirred at 170 °C for 30 min under microwave irradiation. The reaction mixture was concentrated under reduced pressure, and the obtained solid was washed with methanol and collected by filtration. The obtained solid (1.8 g) was mixed with N-methylpyrrolidone (20 mL), (2R)-2-methylpyrrolidine (608 mg), and N,N-diisopropylethylamine (2.5 mL), and the mixture was stirred at 100 °C for 30 min. The reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexaneethyl acetate) to obtain 7 (1.4 g, 37%) as a white solid: 1H-NMR (DMSO-d6) δ: 1.14 (3H, d, J = 6.0 Hz), 1.32–1.44 (1H, m), 1.60–1.72 (2H, m), 1.90–2.00 (1H, m), 2.10–2.20 (1H, m), 2.15 (3H, s), 2.42–2.54 (1H, m), 2.90–3.00 (1H, m), 3.40 (1H, d, J = 14.3 Hz), 4.20 (1H, d, J = 14.3 Hz), 7.66 (1H, d, J = 8.6 Hz), 7.99 (1H, d, J = 10.1 Hz), 8.02 (1H, s), 12.18 (1H, s); ESI-MS m/z 402 (M + H)+.
4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8)A mixture of N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)acetamide (7, 1.4 g), ethanol (10 mL), and a 6 M aqueous sodium hydroxide solution (5 mL) was stirred at 120 °C for 15 min under microwave irradiation. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexaneethyl acetate) to obtain 8 (1.0 g, 83%) as a yellow oil: 1H-NMR (DMSO-d6) δ: 1.11 (3H, d, J = 6.1 Hz), 1.31–1.42 (1H, m), 1.56–1.74 (2H, m), 1.86–2.00 (1H, m), 2.11 (1H, dd, J = 17.5, 8.8 Hz), 2.35–2.46 (1H, m), 2.95–3.03 (1H, m), 3.20 (1H, d, J = 13.9 Hz), 4.02 (1H, d, J = 13.9 Hz), 7.01 (2H, s), 7.58 (1H, d, J = 8.6 Hz), 7.93 (1H, d, J = 9.9 Hz), 7.96 (1H, s); ESI-MS m/z 360 (M + H)+.
6-Chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)pyrimidin-4-amine (9a)Under an argon gas flow, a 60% oil dispersion of sodium hydride (34 mg) was added to a solution of 4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8, 300 mg) and 4,6-dichloropyrimidine (125 mg) in tetrahydrofuran (6.0 mL) while cooling on ice/methanol, and the mixture was stirred at room temperature for 1.5 h. A 60% oil dispersion of sodium hydride (17 mg) was added, and the reaction mixture was stirred at room temperature for 5 h. Ice-cooled water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain 9a (234 mg, 59%) as a pale yellow solid: 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 6.1 Hz), 1.33–1.45 (1H, m), 1.60–1.72 (2H, m), 1.90–2.02 (1H, m), 2.16 (1H, dd, J = 17.4, 8.7 Hz), 2.42–2.54 (1H, m), 2.94–3.04 (1H, m), 3.42 (1H, d, J = 14.3 Hz), 4.23 (1H, d, J = 14.3 Hz), 7.08 (1H, s), 7.68 (1H, d, J = 8.6 Hz), 8.02 (1H, d, J = 9.8 Hz), 8.04 (1H, s), 8.75 (1H, d, J = 0.9 Hz), 12.09 (1H, s); ESI-MS m/z 472, 474 (M + H)+.
4-Chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)pyrimidin-2-amine (9b) 2-Chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)pyrimidin-4-amine (9c)Under an argon gas flow, a 60% oil dispersion of sodium hydride (115 mg) was added to a solution of 4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8, 500 mg) and 2,4-dichloropyrimidine (210 mg) in N-methylpyrrolidone (10 mL) while cooling on ice/methanol, and the mixture was stirred at room temperature for 1 h. 2,4-Dichloropyrimidine (50 mg) and a 60% oil dispersion of sodium hydride (30 mg) were added, and the reaction mixture was stirred at room temperature for 30 min. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane–ethyl acetate) to obtain 9b (71 mg, 11%) and 9c (534 mg, 81%) both as pale yellow solids.
9b: 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 6.1 Hz), 1.33–1.45 (1H, m), 1.60–1.72 (2H, m), 1.90–2.02 (1H, m), 2.16 (1H, dd, J = 17.5, 8.7 Hz), 2.41–2.56 (1H, m), 2.94–3.03 (1H, m), 3.39 (1H, d, J = 14.1 Hz), 4.19 (1H, d, J = 14.1 Hz), 7.20 (1H, d, J = 5.2 Hz), 7.67 (1H, d, J = 8.5 Hz), 8.03 (1H, d, J = 9.8 Hz), 8.07 (1H, s), 8.64 (1H, d, J = 5.2 Hz), 12.11 (1H, s); ESI-MS m/z 472, 474 (M + H)+.
9c: 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 6.0 Hz), 1.33–1.46 (1H, m), 1.60–1.72 (2H, m), 1.89–2.03 (1H, m), 2.18 (1H, dd, J = 17.4, 8.7 Hz), 2.43–2.55 (1H, m), 2.92–3.03 (1H, m), 3.43 (1H, d, J = 14.1 Hz), 4.20 (1H, d, J = 14.1 Hz), 7.04 (1H, d, J = 5.8 Hz), 7.68 (1H, d, J = 8.5 Hz), 8.03–8.10 (2H, m), 8.37 (1H, d, J = 5.8 Hz), 12.23 (1H, s); ESI-MS m/z 472, 474 (M + H)+.
The following compounds (9d–g) were prepared using a procedure similar to that described for 9a.
6-Chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)-2-methylpyrimidin-4-amine (9d)White powder (yield 67%): 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 6.0 Hz), 1.33–1.46 (1H, m), 1.60–1.72 (2H, m), 1.90–2.03 (1H, m), 2.19 (1H, dd, J = 17.5, 8.8 Hz), 2.43–2.53 (1H, m), 2.56 (3H, s), 2.92–3.02 (1H, m), 3.40 (1H, d, J = 14.0 Hz), 4.18 (1H, d, J = 14.0 Hz), 6.90 (1H, s), 7.67 (1H, d, J = 8.6 Hz), 8.04–8.13 (2H, m), 11.99 (1H, s); atmospheric pressure chemical ionization (APCI)/ESI-MS m/z 486 (M + H)+.
6-Chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)-5-methylpyrimidin-4-amine (9e)Pale yellow foam (yield 80%): 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 6.1 Hz), 1.33–1.46 (1H, m), 1.59–1.73 (2H, m), 1.90–2.03 (1H, m), 2.16 (1H, dd, J = 17.5, 8.7 Hz), 2.39 (3H, s), 2.41–2.54 (1H, m), 2.92–3.04 (1H, m), 3.40 (1H, d, J = 14.1 Hz), 4.21 (1H, d, J = 14.1 Hz), 7.67 (1H, d, J = 8.6 Hz), 8.08 (1H, d, J = 9.9 Hz), 8.11 (1H, s), 8.60 (1H, s), 11.28 (1H, s); APCI/ESI-MS m/z 486 (M + H)+.
6-Chloro-5-fluoro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)pyrimidin-4-amine (9f)Yellow solid (yield 108%): 1H-NMR (DMSO-d6) δ: 1.16 (3H, d, J = 6.1 Hz), 1.33–1.46 (1H, m), 1.58–1.73 (2H, m), 1.88–2.02 (1H, m), 2.10–2.24 (1H, m), 2.40–2.54 (1H, m), 2.94–3.05 (1H, m), 3.44 (1H, d, J = 14.0 Hz), 4.24 (1H, d, J = 14.0 Hz), 7.70 (1H, d, J = 8.6 Hz), 8.03 (1H, d, J = 9.8 Hz), 8.07 (1H, s), 8.57 (1H, s), 12.46 (1H, s); APCI/ESI-MS m/z 490 (M + H)+.
6-Chloro-5-fluoro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)-2-methylpyrimidin-4-amine (9g)Yellow solid (yield 94%): 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 5.8 Hz), 1.32–1.46 (1H, m), 1.58–1.72 (2H, m), 1.88–2.02 (1H, m), 2.13–2.27 (1H, m), 2.40–2.54 (1H, m), 2.55 (3H, s), 2.92–3.02 (1H, m), 3.41 (1H, d, J = 14.0 Hz), 4.18 (1H, d, J = 14.0 Hz), 7.67 (1H, d, J = 8.5 Hz), 8.05–8.15 (2H, m), 12.34 (1H, s); APCI/ESI-MS m/z 504 (M + H)+.
3-(4-{6-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyrimidin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3a)To a solution of 6-chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)pyrimidin-4-amine (9a, 230 mg) in N-methylpyrrolidone (3.0 mL) were added ethyl 3-(piperazin-1-yl)propanoate dihydrochloride (380 mg) and N,N-diisopropylethylamine (1.0 mL), followed by stirring at 80 °C for 3 h. The reaction mixture was cooled to room temperature, and water and ethyl acetate were added thereto, followed by extraction with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexanesethyl acetate) to obtain the intermediate (196 mg, 65%) as a yellow syrup. A 1 M aqueous sodium hydroxide solution (1.50 mL) was added to a solution of the obtained intermediate (190 mg) in ethanol (3.0 mL) and tetrahydrofuran (3.0 mL), and the mixture was stirred at 50 °C for 30 min. The reaction mixture was cooled to room temperature, and water and a 1 M aqueous hydrochloric acid solution (3.00 mL) were added, and the mixture was extracted with chloroform/isopropanol, and the organic layer was dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was dissolved in tetrahydrofuran (20 mL), and a 4 M hydrogen chloride/1,4-dioxane solution (2.00 mL) was added thereto. The mixture was concentrated under reduced pressure, and washed with diethyl ether/acetonitrile/water to obtain 3a (88 mg, 41%) as a pale yellow solid: 1H-NMR (DMSO-d6) δ: 1.38 (3H, d, J = 6.5 Hz), 1.59–1.75 (1H, m), 1.82–1.98 (2H, m), 2.11–2.23 (1H, m), 2.89 (2H, t, J = 7.4 Hz), 3.00–3.20 (3H, m), 3.28–3.48 (5H, m), 3.48–3.64 (3H, m), 3.60–4.50 (2H, m), 4.27–4.40 (2H, m), 4.43 (1H, dd, J = 15.0, 7.8 Hz), 4.73 (1H, dd, J = 15.0, 2.2 Hz), 6.33 (1H, s), 7.79 (1H, d, J = 8.7 Hz), 7.88–7.98 (2H, m), 8.47 (1H, d, J = 0.7 Hz), 10.67 (1H, br s), 11.42 (1H, br s), 11.85 (1H, br s); ESI-MS m/z 594 (M + H)+; HRMS (M + H)+ Calcd for C27H32O2N7F4S 594.2269. Found 594.2271. Anal. Calcd for C27H31F4N7O2S·2.9HCl·3H2O·0.1C4H10O: C, 43.25; H, 5.42; N, 12.89; S, 4.21; Cl, 13.51; F, 9.99. Found: C, 43.26; H, 5.53; N, 12.66; S, 4.25; Cl, 13.43; F, 10.00.
The following compounds (3b and 3c) were prepared using a procedure similar to that described for 3a.
3-(4-{2-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyrimidin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3b)Pale yellow solid (yield 19%): 1H-NMR (DMSO-d6) δ: 1.32–1.42 (3H, m), 1.60–1.73 (1H, m), 1.80–1.95 (2H, m), 2.06–2.22 (1H, m), 2.89 (2H, t, J = 7.5 Hz), 2.95–3.09 (1H, m), 3.09–3.26 (2H, m), 3.30–3.76 (8H, m), 3.30–4.90 (3H, m), 4.30–4.42 (1H, m), 4.50–4.90 (3H, m), 6.69 (1H, d, J = 6.6 Hz), 7.80–7.96 (1H, m), 7.85 (1H, d, J = 8.2 Hz), 7.90 (1H, s), 8.19 (1H, d, J = 6.6 Hz), 11.37 (2H, br s); ESI-MS m/z 594 (M + H)+.
3-(4-{4-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyrimidin-2-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3c)White solid (yield 57%): 1H-NMR (DMSO-d6) δ: 1.39 (3H, d, J = 6.5 Hz), 1.61–1.75 (1H, m), 1.80–1.96 (2H, m), 2.07–2.20 (1H, m), 2.87 (2H, t, J = 7.4 Hz), 2.95–3.08 (1H, m), 3.08–3.24 (2H, m), 3.30–3.74 (8H, m), 3.70–5.10 (2H, m), 4.45 (1H, dd, J = 14.9, 7.3 Hz), 4.77 (1H, dd, J = 14.9, 3.3 Hz), 4.77–4.96 (2H, m), 6.45 (1H, d, J = 5.8 Hz), 7.81 (1H, d, J = 8.8 Hz), 7.85–7.94 (2H, m), 8.18 (1H, d, J = 5.8 Hz), 11.18 (1H, br s), 11.54 (1H, br s), 12.22 (1H, br s); ESI-MS m/z 594 (M + H)+; HRMS (M + H)+ Calcd for C27H32O2N7F4S 594.2269. Found 594.2275. Anal. Calcd for C27H31F4N7O2S·3HCl·2H2O: C, 43.88; H, 5.18; N, 13.27; S, 4.34; Cl, 14.39; F, 10.28. Found: C, 43.70; H, 5.41; N, 13.09; S, 4.35; Cl, 14.32; F, 10.42.
3-(4-{6-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-2-methylpyrimidin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3d)6-Chloro-N-(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)-2-methylpyrimidin-4-amine (9d, 453 mg), N,N-diisopropylethylamine (2.00 mL), ethyl 3-(piperazin-1-yl)propanoate dihydrochloride (750 mg), and N-methylpyrrolidone (10 mL) were mixed, followed by stirring at 80 °C for 1 h. N,N-Diisopropylethylamine (2.00 mL) and ethyl 3-(piperazin-1-yl)propanoate dihydrochloride (750 mg) were added, and the mixture was stirred at 80 °C overnight. The reaction mixture was cooled to room temperature, and water and ethyl acetate were added thereto, followed by extraction with ethyl acetate. The organic layer was washed with water and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexanesethyl acetate).
To a solution of the obtained residue in ethanol (5.0 mL) and tetrahydrofuran (5.0 mL) was added a 1 M aqueous sodium hydroxide solution (5.00 mL), followed by stirring at 60 °C for 1 h. The reaction mixture was cooled to room temperature, and a 1 M aqueous hydrochloric acid solution (5.00 mL) was added thereto, followed by extraction with chloroform/methanol. The organic layer was dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (chloroform-methanol).
The obtained residue was mixed with ethyl acetate, a 4 M hydrogen chloride/1,4-dioxane solution (700 µL) was added, and the mixture was concentrated under reduced pressure, and washed with ethyl acetate to obtain 3d (525 mg, 79%) as a white powder: 1H-NMR (DMSO-d6) δ: 1.38 (3H, d, J = 6.4 Hz), 1.59–1.75 (1H, m), 1.82–1.98 (2H, m), 2.10–2.23 (1H, m), 2.48 (3H, s), 2.88 (2H, t, J = 7.3 Hz), 2.98–3.18 (3H, m), 3.26–3.47 (5H, m), 3.47–3.63 (3H, m), 3.50–4.50 (2H, m), 4.25–4.40 (2H, m), 4.43 (1H, dd, J = 14.9, 7.7 Hz), 4.74 (1H, dd, J = 14.9, 2.4 Hz), 6.14 (1H, s), 7.79 (1H, d, J = 8.7 Hz), 7.87–7.97 (2H, m), 10.65 (1H, br s), 11.26 (1H, br s), 11.75 (1H, br s); ESI-MS m/z 608 (M + H)+; HRMS (M + H)+ Calcd for C28H34O2N7F4S 608.2425. Found 608.2419. Anal. Calcd for C28H33F4N7O2S·2.8HCl·3H2O: C, 44.03; H, 5.52; N, 12.84; S, 4.20; Cl, 13.00; F, 9.95. Found: C, 44.34; H, 5.58; N, 12.63; S, 4.19; Cl, 13.05; F, 9.96.
The following compound (3e) was prepared using a procedure similar to that described for 3d.
3-(4-{6-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-5-methylpyrimidin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3e)Off-white powder (yield 9.2%): 1H-NMR (DMSO-d6) δ: 1.37 (3H, d, J = 6.4 Hz), 1.58–1.75 (1H, m), 1.83–1.97 (2H, m), 2.11–2.24 (1H, m), 2.19 (3H, s), 2.89 (2H, t, J = 7.4 Hz), 2.97–3.25 (3H, m), 3.29–3.47 (5H, m), 3.47–3.59 (3H, m), 3.60–4.80 (2H, m), 3.74–3.88 (2H, m), 4.44 (1H, dd, J = 15.1, 7.9 Hz), 4.75 (1H, dd, J = 15.1, 2.3 Hz), 7.80 (1H, d, J = 8.7 Hz), 7.91–8.02 (2H, m), 8.51 (1H, s), 10.60 (1H, br s), 11.16–11.38 (2H, m); ESI-MS m/z 608 (M + H)+; HRMS (M + H)+ Calcd for C28H34O2N7F4S 608.2425. Found 608.2423. Anal. Calcd for C28H33F4N7O2S·2.8HCl·3H2O: C, 44.03; H, 5.52; N, 12.84; S, 4.20; Cl, 13.00; F, 9.95. Found: C, 44.15; H, 5.59; N, 12.47; S, 4.15; Cl, 13.09; F, 9.86.
The following compounds (3f and 3g) were prepared using a procedure similar to that described for 3a.
3-(4-{5-Fluoro-6-[(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyrimidin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3f)White powder (yield 34%): 1H-NMR (DMSO-d6) δ: 1.38 (3H, d, J = 6.5 Hz), 1.59–1.76 (1H, m), 1.82–1.99 (2H, m), 2.10–2.25 (1H, m), 2.88 (2H, t, J = 7.3 Hz), 3.07–3.24 (3H, m), 3.26–3.36 (2H, m), 3.36–3.48 (1H, m), 3.48–3.66 (5H, m), 3.70–4.70 (2H, m), 4.36–4.54 (3H, m), 4.75 (1H, dd, J = 14.8, 2.0 Hz), 7.80 (1H, d, J = 8.7 Hz), 7.91–8.02 (2H, m), 8.31 (1H, d, J = 1.4 Hz), 10.73 (1H, br s), 11.45 (1H, br s), 12.01 (1H, br s); ESI-MS m/z 612 (M + H)+; HRMS (M + H)+ Calcd for C27H31O2N7F5S 612.2175. Found 612.2175. Anal. Calcd for C27H30F5N7O2S·3HCl·2H2O: C, 42.84; H, 4.93; N, 12.95; S, 4.24; Cl, 14.05; F, 12.55. Found: C, 42.77; H, 4.88; N, 12.91; S, 4.22; Cl, 14.09; F, 12.69.
3-(4-{5-Fluoro-6-[(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-2-methylpyrimidin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3g)White powder (yield 31%): 1H-NMR (DMSO-d6) δ: 1.39 (3H, d, J = 6.5 Hz), 1.61–1.75 (1H, m), 1.84–1.98 (2H, m), 2.10–2.23 (1H, m), 2.48 (3H, s), 2.88 (2H, t, J = 7.3 Hz), 3.05–3.21 (3H, m), 3.26–3.35 (2H, m), 3.35–3.47 (1H, m), 3.46–3.62 (5H, m), 3.90–4.90 (2H, m), 4.35–4.50 (3H, m), 4.74 (1H, dd, J = 14.9, 2.3 Hz), 7.79 (1H, d, J = 8.7 Hz), 7.89–7.99 (2H, m), 10.91 (1H, br s), 11.44 (1H, br s), 11.90 (1H, br s); ESI-MS m/z 626 (M + H)+; HRMS (M + H)+ Calcd for C28H33O2N7F5S 626.2331. Found 626.2330. Anal. Calcd for C28H32F5N7O2S·3HCl·1.5H2O: C, 44.13; H, 5.03; N, 12.87; S, 4.21; Cl, 13.96; F, 12.47. Found: C, 44.00; H, 5.01; N, 12.76; S, 4.19; Cl, 13.95; F, 12.49.
Ethyl 3-[4-(4-Chloro-1,3,5-triazin-2-yl)piperazin-1-yl]propanoate (11h)To a solution of 2,4-dichloro-1,3,5-triazine (10h, 1.00 g) in N-methylpyrrolidone (20 mL) was added dipotassium carbonate (2.77 g), and ethyl 3-(piperazin-1-yl)propanoate dihydrochloride (1.73 g) while cooling on ice/methanol, followed by stirring at the same temperature for 1 h. The reaction mixture was warmed to room temperature and stirred for 1.5 h. Water and ethyl acetate were added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane-ethyl acetate) to obtain 11h (1.32 g, 66%) as a colorless oil: 1H-NMR (DMSO-d6) δ: 1.19 (3H, d, J = 7.1 Hz), 2.42–2.52 (6H, m), 2.62 (2H, t, J = 6.9 Hz), 3.67–3.74 (2H, m), 3.74–3.82 (2H, m), 4.07 (2H, q, J = 7.1 Hz), 8.47 (1H, s); ESI-MS m/z 300, 302 (M + H)+.
Ethyl 3-[4-(6-Chloropyridazin-3-yl)piperazin-1-yl]propanoate (11i)3,6-Dichloropyridazine (10i, 1.50 g), ethyl 3-(piperazin-1-yl)propanoate dihydrochloride (5.00 g), dipotassium carbonate (8.50 g), and N-methylpyrrolidone (30 mL) were mixed, followed by stirring at 80 °C overnight. The reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and a saturated aqueous sodium chloride solution, and dried over anhydrous magnesium sulfate. The insoluble materials were then separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography (chloroform-ethyl acetate), and washed with hexanes/diethyl ether to obtain 11i (2.04 g, 68%) as a light yellow solid: 1H-NMR (DMSO-d6) δ: 1.19 (3H, d, J = 7.1 Hz), 2.46–2.60 (6H, m), 2.62–2.74 (2H, m), 3.51–3.64 (4H, m), 4.07 (2H, q, J = 7.1 Hz), 7.38 (1H, d, J = 9.7 Hz), 7.52 (1H, d, J = 9.7 Hz); ESI-MS m/z 299 (M + H)+.
The following compounds (11j and 11k–m) were prepared using a procedure similar to that described for 11i.
Ethyl 3-[4-(6-Chloropyrazin-2-yl)piperazin-1-yl]propanoate (11j)Colorless oil (yield 69%): 1H-NMR (DMSO-d6) δ: 1.19 (3H, d, J = 7.1 Hz), 2.44–2.53 (6H, m), 2.62 (2H, t, J = 6.8 Hz), 3.51–3.58 (4H, m), 4.07 (2H, q, J = 7.1 Hz), 7.84 (1H, s), 8.27 (1H, s); ESI-MS m/z 299 (M + H)+.
Ethyl 3-[4-(6-Bromopyridin-2-yl)piperazin-1-yl]propanoate (11k)Colorless oil (yield 87%): 1H-NMR (DMSO-d6) δ: 1.19 (3H, d, J = 7.1 Hz), 2.42–2.48 (4H, m), 2.48–2.53 (2H, m), 2.57–2.64 (2H, m), 3.40–3.48 (4H, m), 4.07 (2H, q, J = 7.1 Hz), 6.64 (1H, d, J = 7.3 Hz), 6.76 (1H, d, J = 8.4 Hz), 7.53 (1H, dd, J = 8.4, 7.3 Hz); ESI-MS m/z 342, 344 (M + H)+.
Ethyl 3-[4-(4-Chloropyridin-2-yl)piperazin-1-yl]propanoate (11l)Colorless oil (yield 17%): 1H-NMR (DMSO-d6) δ: 1.18 (3H, d, J = 7.1 Hz), 2.41–2.46 (4H, m), 2.46–2.52 (2H, m), 2.57–2.64 (2H, m), 3.44–3.51 (4H, m), 4.07 (2H, q, J = 7.1 Hz), 6.69 (1H, dd, J = 5.3, 1.6 Hz), 6.90 (1H, d, J = 1.6 Hz), 8.05 (1H, d, J = 5.3 Hz); ESI-MS m/z 298, 300 (M + H)+.
Ethyl 3-[4-(2-Chloropyridin-4-yl)piperazin-1-yl]propanoate (11m)Colorless oil (yield 72%): 1H-NMR (DMSO-d6) δ: 1.18 (3H, d, J = 7.1 Hz), 2.42–2.52 (6H, m), 2.57–2.64 (2H, m), 3.30–3.36 (4H, m), 4.07 (2H, q, J = 7.1 Hz), 6.80–6.86 (2H, m), 7.91–7.96 (1H, m); ESI-MS m/z 298, 300 (M + H)+.
Ethyl 3-[4-(3-Bromophenyl)piperazin-1-yl]propanoate (11n)1-(3-Bromophenyl)piperazine (12, 2.00 g), ethyl acrylate (2.70 mL), and ethanol (6.0 mL) were mixed, followed by heating to reflux for 6 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to obtain 11n (3.14 g, 111%) as a colorless syrup: 1H-NMR (DMSO-d6) δ: 1.18 (3H, d, J = 7.1 Hz), 2.45–2.54 (6H, m), 2.61 (2H, t, J = 7.4 Hz), 3.08–3.18 (4H, m), 4.07 (2H, q, J = 7.1 Hz), 6.87–6.95 (2H, m), 7.03–7.07 (1H, m), 7.13 (1H, t, d, J = 8.1 Hz); APCI/ESI-MS m/z 341,343 (M + H)+.
3-(4-{4-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-1,3,5-triazin-2-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3h)Under an argon gas flow, a 60% oil dispersion of sodium hydride (110 mg) was added to a solution of 4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8, 335 mg) and ethyl 3-[4-(4-chloro-1,3,5-triazin-2-yl)piperazin-1-yl]propanoate (11h, 335 mg) in tetrahydrofuran (7.0 mL) while cooling on ice/methanol, and the mixture was stirred at 0 °C for 1.5 h. Water was added to the reaction mixture and the mixture was extracted with ethyl acetate. The organic layer was washed with a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography (hexane-ethyl acetate) to obtain the intermediate (396 mg, 68%) as a white solid.
To a suspension of the obtained intermediate (374 mg) in ethanol (5.0 mL) and tetrahydrofuran (5.0 mL) was added a 1 M aqueous sodium hydroxide solution (4.00 mL), followed by stirring at 50 °C for 30 min. The reaction mixture was cooled to room temperature, water (20 mL) and a 1 M aqueous hydrochloric acid solution (4.00 mL) were added, and the mixture was extracted with chloroform/isopropanol, and the organic layer was dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure.
The residue was dissolved in tetrahydrofuran (10 mL), and a 4 M hydrogen chloride/1,4-dioxane solution (2.0 mL) was added thereto. The mixture was concentrated under reduced pressure, and washed with acetonitrile and water to obtain 3h (324 mg, 77%) as a pale yellow solid: 1H-NMR (DMSO-d6) δ: 1.39 (3H, s), 1.60–1.76 (1H, m), 1.78–1.98 (2H, m), 2.04–2.22 (1H, m), 2.80–2.96 (2H, m), 2.96–3.26 (3H, m), 3.26–3.85 (8H, m), 4.00–5.50 (2H, m), 4.34–4.51 (1H, m), 4.64–4.85 (2H, m), 4.89–5.13 (1H, m), 7.81 (1H, d, J = 8.7 Hz), 7.82–7.95 (2H, m), 8.48 (1H, s), 11.62 (1H, br s), 11.78 (1H, br s), 12.16 (1H, br s); ESI-MS m/z 595 (M + H)+; HRMS (M + H)+ Calcd for C26H31O2N8F4S 595.2221. Found 595.2218. Anal. Calcd for C26H30F4N8O2S·2.8HCl·1.75H2O: C, 42.88; H, 5.02; N, 15.39; S, 4.40; Cl, 13.63; F, 10.44. Found: C, 42.82; H, 5.25; N, 15.25; S, 4.26; Cl, 13.89; F, 10.52.
3-(4-{6-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyridazin-3-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3i)4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8, 500 mg), ethyl 3-[4-(6-chloropyridazin-3-yl)piperazin-1-yl]propanoate (11i, 520 mg), tris(dibenzylideneacetone)dipalladium(0) (91 mg), di-tert-butyl(2′,4′,6′-triisopropylbiphenyl-2-yl)phosphine (96 mg), cesium carbonate (970 mg), toluene (10 mL), and water (1.0 mL) were mixed, followed by stirring at 80 °C for 7 h. Tris(dibenzylideneacetone)dipalladium(0) (100 mg), di-tert-butyl(2′,4′,6′-triisopropylbiphenyl-2-yl)phosphine (100 mg), and cesium carbonate (1.00 g) were added, and the mixture was stirred at 100 °C for 6 h. The reaction mixture was cooled to room temperature, diluted with water and ethyl acetate, and filtered through a pad of Celite™. The filtrate was separated, and the organic layer was concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography (hexanes-ethyl acetate) and then by silica gel column chromatography (chloroform-ethyl acetate) to obtain the intermediate (172 mg, 20%) as a yellow oil.
To a solution of the obtained intermediate (172 mg) in ethanol (3.0 mL) and tetrahydrofuran (3.0 mL) was added a 1 M aqueous sodium hydroxide solution (1.60 mL), followed by stirring at 80 °C for 2 h. The reaction mixture was cooled to room temperature, and a 1 M aqueous hydrochloric acid solution (1.60 mL) was added thereto, followed by extraction with chloroform/methanol. The organic layer was dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was mixed with ethyl acetate, and a 4 M hydrogen chloride/1,4-dioxane solution (250 µL) was added thereto. The mixture was concentrated under reduced pressure, and washed with ethyl acetate to obtain 3i (97 mg, 50%) as a yellow powder: 1H-NMR (DMSO-d6) δ: 1.39 (3H, d, J = 6.4 Hz), 1.60–1.76 (1H, m), 1.84–1.98 (2H, m), 2.11–2.24 (1H, m), 2.92 (2H, t, J = 7.4 Hz), 3.06–3.25 (3H, m), 3.28–3.66 (8H, m), 4.00–5.50 (2H, m), 4.27–4.40 (2H, m), 4.45 (1H, dd, J = 15.0, 7.7 Hz), 4.74 (1H, dd, J = 15.0, 1.9 Hz), 7.40 (1H, d, J = 9.7 Hz), 7.66 (1H, d, J = 9.7 Hz), 7.79 (1H, d, J = 8.6 Hz), 7.90–8.00 (2H, m), 10.78 (1H, br s), 11.49 (1H, br s), 11.85 (1H, br s); ESI-MS m/z 594 (M + H)+; HRMS (M + H)+ Calcd for C27H32O2N7F4S 594.2269. Found 594.2276. Anal. Calcd for C27H31F4N7O2S·3HCl·3H2O: C, 42.83; H, 5.33; N, 12.95; S, 4.24; Cl, 14.05; F, 10.04. Found: C, 43.42; H, 5.41; N, 12.05; S, 4.02; Cl, 13.58; F, 9.63.
The following compounds (3j, and 3k–n) were prepared using a procedure similar to that described for 3i.
3-(4-{6-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyrazin-2-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3j)Yellow solid (yield 6.5%): 1H-NMR (DMSO-d6) δ: 1.39 (3H, d, J = 6.4 Hz), 1.61–1.74 (1H, m), 1.80–1.95 (2H, m), 2.06–2.20 (1H, m), 2.88 (2H, t, J = 7.4 Hz), 2.97–3.10 (1H, m), 3.10–3.26 (2H, m), 3.30–3.56 (6H, m), 3.40–4.20 (2H, m), 3.62–3.72 (2H, m), 4.44 (1H, dd, J = 14.8, 7.4 Hz), 4.52–4.64 (2H, m), 4.75 (1H, dd, J = 14.9, 2.9 Hz), 7.77–7.84 (2H, m), 7.85–7.92 (2H, m), 7.95 (1H, s), 11.07 (1H, br s), 11.31 (1H, br s), 11.90 (1H, br s); ESI-MS m/z 594 (M + H)+; HRMS (M + H)+ Calcd for C27H32O2N7F4S 594.2269. Found 594.2273. Anal. Calcd for C27H31F4N7O2S·2.6HCl·3.5H2O: C, 43.15; H, 5.45; N, 13.05; S, 4.27; Cl, 12.27; F, 10.11. Found: C, 43.20; H, 5.40; N, 12.65; S, 4.21; Cl, 12.55; F, 9.83.
3-(4-{6-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyridin-2-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3k)White solid (yield 33%): 1H-NMR (DMSO-d6) δ: 1.40 (3H, d, J = 6.5 Hz), 1.62–1.76 (1H, m), 1.81–1.99 (2H, m), 2.08–2.22 (1H, m), 2.91 (2H, t, J = 8.2 Hz), 3.02–3.23 (3H, m), 3.29–3.56 (6H, m), 3.57–3.73 (2H, m), 4.20–6.00 (2H, m), 4.43 (1H, dd, J = 15.0, 7.3 Hz), 4.43–4.59 (2H, m), 4.71 (1H, dd, J = 15.0, 3.0 Hz), 6.45 (1H, d, J = 7.8 Hz), 6.50 (1H, d, J = 8.3 Hz), 7.54–7.60 (1H, m), 7.78 (1H, d, J = 8.7 Hz), 7.86–7.95 (2H, m), 11.43 (2H, br s), 11.53 (1H, br s); ESI-MS m/z 593 (M + H)+; HRMS (M + H)+ Calcd for C28H33O2N6F4S 593.2316. Found 593.2319. Anal. Calcd for C28H32F4N6O2S·2.5HCl·2H2O: C, 46.72; H, 5.39; N, 11.67 S, 4.45 Cl, 12.31; F, 10.56. Found: C, 46.90; H, 5.51; N, 11.40; S, 4.34; Cl, 12.51; F, 10.48.
3-(4-{4-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyridin-2-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3l)Light brown solid (yield 78%): 1H-NMR (DMSO-d6) δ: 1.41 (3H, d, J = 6.4 Hz), 1.61–1.76 (1H, m), 1.82–1.98 (2H, m), 2.10–2.23 (1H, m), 2.88 (2H, t, J = 7.4 Hz), 3.00–4.50 (12H, m), 3.01–3.15 (1H, m), 4.15–4.40 (2H, m), 4.46 (1H, dd, J = 14.7, 7.1 Hz), 4.78 (1H, d, J = 14.7 Hz), 7.13–7.27 (1H, m), 7.76–7.89 (2H, m), 7.97–8.08 (3H, m), 11.23 (1H, br s), 11.53 (1H, br s), 12.68 (1H, br s); ESI-MS m/z 593 (M + H)+; HRMS (M + H)+ Calcd for C28H33O2N6F4S 593.2316. Found 593.2323. Anal. Calcd for C28H32F4N6O2S·3HCl·2H2O: C, 45.57; H, 5.33; N, 11.39; S, 4.34 Cl, 14.41; F, 10.30. Found: C, 45.33; H, 5.51; N, 11.37; S, 4.36; Cl, 14.58; F, 10.22.
3-(4-{2-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyridin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3m)White solid (yield 26%): 1H-NMR (DMSO-d6) δ: 1.38 (3H, d, J = 6.4 Hz), 1.60–1.75 (1H, m), 1.83–1.98 (2H, m), 2.11–2.23 (1H, m), 2.90 (2H, t, J = 7.4 Hz), 3.04–3.27 (3H, m), 3.30–3.70 (8H, m), 3.80–5.10 (3H, m), 3.95–4.18 (2H, m), 4.41 (1H, dd, J = 14.9, 7.7 Hz), 4.71 (1H, dd, J = 14.9, 1.8 Hz), 6.56–7.00 (2H, m), 7.80 (1H, d, J = 8.6 Hz), 7.92–8.04 (2H, m), 8.07 (1H, d, J = 6.8 Hz), 10.90 (1H, br s), 11.59 (1H, br s); ESI-MS m/z 593 (M + H)+; HRMS (M + H)+ Calcd for C28H33O2N6F4S 593.2316. Found 593.2314. Anal. Calcd for C28H32F4N6O2S·3.1HCl·2.5H2O: C, 44.80; H, 5.38; N, 11.19; S, 4.27; Cl, 14.64; F, 10.12. Found: C, 44.67; H, 5.53; N, 11.32; S, 4.35; Cl, 14.45; F, 10.24.
3-(4-{3-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]phenyl}piperazin-1-yl)propanoic Acid Trihydrochloride (3n)Pale yellow powder (yield 33%): 1H-NMR (DMSO-d6) δ: 1.38 (3H, d, J = 6.4 Hz), 1.60–1.73 (1H, m), 1.82–1.96 (2H, m), 2.10–2.22 (1H, m), 2.88 (2H, t, J = 7.4 Hz), 3.00–3.25 (5H, m), 3.30–3.45 (3H, m), 3.45–3.60 (3H, m), 3.60–4.30 (2H, m), 3.70–3.84 (2H, m), 4.35 (1H, dd, J = 15.0, 7.7 Hz), 4.69 (1H, dd, J = 15.0, 2.4 Hz), 6.68 (1H, dd, J = 8.1, 1.9 Hz), 7.08 (1H, dd, J = 8.1, 1.4 Hz), 7.22 (1H, t, J = 8.1 Hz), 7.35–7.42 (1H, m), 7.79 (1H, d, J = 8.6 Hz), 7.87–7.95 (2H, m), 10.73 (1H, br s), 10.87 (1H, br s), 11.03 (1H, br s); ESI-MS m/z 592 (M + H)+; HRMS (M + H)+ Calcd for C29H34O2N5F4S 592.2364. Found 592.2360. Anal. Calcd for C29H33F4N5O2S·2.5HCl·3H2O: C, 47.27; H, 5.68; N, 9.50; S, 4.35; Cl, 12.03; F, 10.31. Found: C, 47.97; H, 5.59; N, 9.29; S, 4.35; Cl, 11.65; F, 10.15.
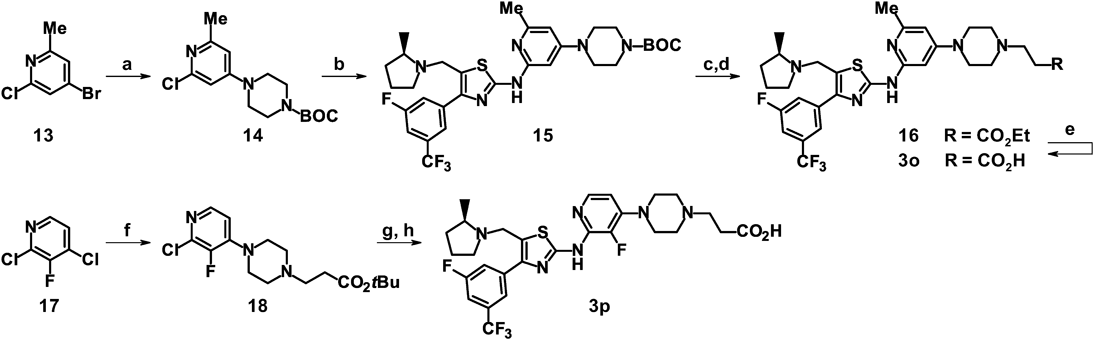
tert-Butyl 4-(2-Chloro-6-methylpyridin-4-yl)piperazine-1-carboxylate (14)4-Bromo-2-chloro-6-methylpyridine (13, 1.70 g), tert-butyl piperazine-1-carboxylate (1.30 g), tris(dibenzylideneacetone)dipalladium(0) (150 mg), (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphine) (285 mg), sodium 2-methylpropan-2-olate (1.00 g), and toluene (30 mL) were mixed, followed by stirring at 100 °C for 2 h. The reaction mixture was cooled to room temperature, diluted with water and ethyl acetate, and filtered through a pad of Celite™. The filtrate was separated, and the organic layer was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexanes-ethyl acetate) to obtain 14 (1.89 g, 87%) as a yellow solid: 1H-NMR (DMSO-d6) δ: 1.42 (9H, s), 2.29 (3H, s), 3.33–3.38 (4H, m), 3.38–3.44 (4H, m), 6.68 (1H, d, J = 2.0 Hz), 6.71 (1H, d, J = 2.0 Hz); ESI-MS m/z 312, 314 (M + H)+.
tert-Butyl 4-{2-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-6-methylpyridin-4-yl}piperazine-1-carboxylate (15)tert-Butyl 4-(2-Chloro-6-methylpyridin-4-yl)piperazine-1-carboxylate (14, 180 mg), 4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8, 179 mg), tris(dibenzylideneacetone)dipalladium(0) (230 mg), 1,1′-binaphthalene-2,2′-diylbis(diphenylphosphine) (330 mg), cesium carbonate (660 mg), and N-methylpyrrolidone (6.0 mL) were mixed, followed by stirring at 100 °C for 6 h. The reaction mixture was cooled to room temperature, diluted with water and ethyl acetate, and filtered through a pad of Celite™. The filtrate was separated, and the organic layer was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (chloroform-ethyl acetate), and purified by silica gel column chromatography (hexanes-ethyl acetate) to obtain 15 (156 mg, 49%) as a pale yellow solid: 1H-NMR (DMSO-d6) δ: 1.16 (3H, d, J = 6.0 Hz), 1.33–1.46 (1H, m), 1.43 (9H, s), 1.59–1.71 (2H, m), 1.90–2.02 (1H, m), 2.18 (1H, dd, J = 17.6, 8.9 Hz), 2.35 (3H, s), 2.40–2.53 (1H, m), 2.92–3.02 (1H, m), 3.21–3.40 (5H, m), 3.40–3.50 (4H, m), 4.11 (1H, d, J = 13.8 Hz), 6.30 (1H, d, J = 1.6 Hz), 6.43 (1H, d, J = 1.6 Hz), 7.62 (1H, d, J = 8.5 Hz), 8.07–8.15 (2H, m), 10.93 (1H, s); APCI/ESI-MS m/z 635 (M + H)+.
Ethyl 3-(4-{2-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-6-methylpyridin-4-yl}piperazin-1-yl)propanoate (16)tert-Butyl 4-{2-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-6-methylpyridin-4-yl}piperazine-1-carboxylate (15, 1.03 g), trifluoroacetic acid (15 mL), and dichloromethane (15 mL) were mixed, followed by stirring at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure, diluted with chloroform, washed with a saturated aqueous sodium hydrogen carbonate solution, and dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography (chloroform–methanol) to obtain the intermediate (600 mg, 69%) as a beige solid. The obtained intermediate (600 mg) was mixed with ethyl acrylate (250 µL) and ethanol (10 mL), and the mixture was stirred at 100 °C under microwave irradiation. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by basic silica gel column chromatography (hexanes–ethyl acetate) to obtain 16 (478 mg, 67%) as a pale yellow powder: 1H-NMR (DMSO-d6) δ: 1.16 (3H, d, J = 6.0 Hz), 1.19 (3H, t, J = 7.1 Hz), 1.33–1.46 (1H, m), 1.58–1.72 (2H, m), 1.90–2.03 (1H, m), 2.18 (1H, dd, J = 17.6, 8.8 Hz), 2.34 (3H, s), 2.39–2.56 (7H, m), 2.61 (2H, t, J = 7.2 Hz), 2.92–3.02 (1H, m), 3.17–3.26 (4H, m), 3.26–3.36 (1H, m), 4.07 (2H, q, J = 7.1 Hz), 4.11 (1H, d, J = 13.8 Hz), 6.26 (1H, d, J = 1.5 Hz), 6.42 (1H, d, J = 1.5 Hz), 7.62 (1H, d, J = 8.6 Hz), 8.08–8.17 (2H, m), 10.91 (1H, s); APCI/ESI-MS m/z 635 (M + H)+.
Sodium 3-(4-{2-[(4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-6-methylpyridin-4-yl}piperazin-1-yl)propanoate (3o)A mixture of ethyl 3-(4-{2-[(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]-6-methylpyridin-4-yl}piperazin-1-yl)propanoate (16, 478 mg) and a 1 M aqueous sodium hydroxide solution (4.5 mL) in tetrahydrofuran (10 mL) and ethanol (10 mL) was stirred at 60 °C for 30 min. The reaction mixture was cooled to room temperature, and a 1 M aqueous hydrochloric acid solution (4.5 mL) was added thereto. The mixture was extracted with chloroform/isopropanol, and the organic layer was dried over anhydrous magnesium sulfate. The insoluble materials were separated by filtration and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (chloroform–methanol) to give a white solid (458 mg). The solid was mixed with ethyl acetate, and a 4 M hydrogen chloride/ethyl acetate solution (1.0 mL) was added thereto. The mixture was concentrated under reduced pressure, and washed with ethyl acetate to give a white solid (393 mg). A 1 M aqueous sodium hydroxide solution (800 µL) was added to the solid and the mixture was purified by reversed-phase column chromatography (acetonitrile–water) to give 3o (164 mg, 35%) as a pale yellow powder: 1H-NMR (DMSO-d6) δ: 1.15 (3H, d, J = 6.0 Hz), 1.33–1.46 (1H, m), 1.58–1.71 (2H, m), 1.90–2.02 (1H, m), 2.12–2.26 (3H, m), 2.34 (3H, s), 2.38–2.64 (7H, m), 2.92–3.02 (1H, m), 3.16–3.26 (4H, m), 3.30 (1H, d, J = 13.7 Hz), 4.10 (1H, d, J = 13.7 Hz), 6.26 (1H, d, J = 1.4 Hz), 6.41 (1H, d, J = 1.4 Hz), 7.61 (1H, d, J = 8.5 Hz), 8.07–8.16 (2H, m), 10.91 (1H, br s); ESI-MS m/z 607 (M + H)+; HRMS (M + H)+ Calcd for C29H35O2N6F4S 607.2473. Found 607.2480. Anal. Calcd for C29H33F4N6O2S·Na·3.5H2O: C, 50.35; H, 5.83; N, 12.15; S, 4.64; F, 10.99; Na, 3.32. Found: C, 50.99; H, 5.78; N, 11.46; S, 4.44; F, 10.44; Na, 3.24.
The following compound (18) was prepared using a procedure similar to that described for 11i.
tert-Butyl 3-[4-(2-Chloro-3-fluoropyridin-4-yl)piperazin-1-yl]propanoate (18)Pale yellow solid (yield 81%): 1H-NMR (DMSO-d6) δ: 1.40 (9H, s), 2.37 (2H, t, J = 7.2 Hz), 2.48–2.54 (4H, m), 2.58 (2H, t, J = 7.2 Hz), 3.24–3.31 (4H, m), 7.01 (1H, dd, J = 6.7, 5.6 Hz), 7.93 (1H, d, J = 5.6 Hz); ESI-MS m/z 344, 346 (M + H)+.
3-(4-{3-Fluoro-2-[(4-[3-fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-yl)amino]pyridin-4-yl}piperazin-1-yl)propanoic Acid Trihydrochloride (3p)4-[3-Fluoro-5-(trifluoromethyl)phenyl]-5-{[(2R)-2-methylpyrrolidin-1-yl]methyl}-1,3-thiazol-2-amine (8, 500 mg), tert-butyl 3-[4-(2-chloro-3-fluoropyridin-4-yl)piperazin-1-yl]propanoate (18, 497 mg), tris(dibenzylideneacetone)dipalladium(0) (640 mg), 1,1′-binaphthalene-2,2′-diylbis(diphenylphosphine) (900 mg), cesium carbonate (1.90 g), and N-methylpyrrolidone (15 mL) were mixed, followed by stirring at 100 °C for 6 h. The reaction mixture was cooled to room temperature, diluted with water and ethyl acetate, and filtered through a pad of Celite™. The filtrate was separated, and the organic layer was washed with water and a saturated aqueous sodium chloride solution. The residue was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography three times (chloroform-ethyl acetate, hexanes-ethyl acetate, and then chloroform-ethyl acetate) to obtain the intermediate (387 mg, 42%) as a brown foam. The obtained intermediate (387 mg) and a 4 M hydrogen chloride/1,4-dioxane solution (18.0 mL) were mixed and then stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and the residue was washed with ethyl acetate to obtain 3p (394 mg, 94%) as a beige solid: 1H-NMR (DMSO-d6) δ: 1.38 (3H, d, J = 6.4 Hz), 1.59–1.75 (1H, m), 1.82–1.98 (2H, m), 2.10–2.24 (1H, m), 2.91 (2H, t, J = 7.4 Hz), 3.07–3.28 (3H, m), 3.10–4.80 (2H, m), 3.30–3.49 (5H, m), 3.49–3.63 (3H, m), 3.75–3.90 (2H, m), 4.43 (1H, dd, J = 15.0, 7.8 Hz), 4.72 (1H, dd, J = 15.0, 2.2 Hz), 6.80 (1H, t, J = 6.1 Hz), 7.79 (1H, d, J = 8.7 Hz), 7.91–8.03 (3H, m), 10.70 (1H, br s), 11.30–11.80 (1H, m), 11.52 (1H, br s); ESI-MS m/z 611 (M + H)+; HRMS (M + H)+ Calcd for C28H32O2N6F5S 611.2222. Found 611.2224. Anal. Calcd for C28H31F5N6O2S·2.9HCl·3H2O: C, 43.65; H, 5.22; N, 10.91; S, 4.16; Cl, 13.35; F, 12.33. Found: C, 43.71; H, 5.32; N, 10.30; S, 4.04; Cl, 13.49; F, 12.02.