Abstract
Peptide bond formation in living cells occurs at the peptidyl transferase center (PTC) of the large ribosomal subunit and involves the transfer of the peptidyl group from peptidyl-tRNA to aminoacyl-tRNA. Despite numerous kinetic and theoretical studies, many details of this reaction —such as whether it proceeds via a stepwise or concerted mechanism— remain unclear. In this study, we calculated the geometry and energy of the transition states and intermediates in peptide bond formation in the PTC environment using the ONIOM (our own n-layered integrated molecular orbital and molecular mechanics) method. The calculations indicated that the energy of the transition states of stepwise mechanisms are lower than those of concerted mechanisms and suggested that the reaction involves a neutral tetrahedral intermediate that is stabilized through the hydrogen-bonding network in the PTC environment. The results will lead to a better understanding of the mechanism of peptidyl transfer reaction, and resolve fundamental questions of the steps and molecular intermediates involved in peptide bond formation in the ribosome.
Introduction
We report here the calculation of the geometry and energy of the transition states and intermediates in peptide bond formation in the peptidyl transferase center (PTC) environment using the ONIOM (our own n-layered integrated molecular orbital and molecular mechanics) method. This approach allowed us to consider electrostatic and steric effects on intermediates and transition states of peptide bond formation proceeding in the PTC under steric constraints imposed by factors outside the system.
The final step in the flow of genetic information from mRNA to protein is peptide bond synthesis on the ribosome.1) Peptide bond formation occurs at the PTC on the large subunit of the ribosome and involves the transfer of the peptidyl group of peptidyl-tRNA to the amino acid residue of aminoacyl- tRNA.2,3)
Despite extensive kinetic and theoretical investigations, the mechanistic details of the peptidyl transfer reaction remain unclear.4) Recent model calculations have provided various ideas and concepts for the mechanism of the peptidyl transfer reaction, and calculations using quantum mechanical/molecular mechanical (QM/MM) methods are expected to provide a deeper understanding of the mechanism of protein synthesis in the enzymatic environment. Further, experimental approaches such as X-ray crystallography and kinetic analyses have provided insights into the basic aspects of peptide bond formation at the PTC. The three-dimensional crystal structure of the 50S (large) ribosomal subunit of Haloarcula marismortui revealed the arrangement of aminoacyl-tRNA and peptidyl-tRNA at the PTC at atomic-level resolution.5) Moreover, a kinetic study of ribosomal peptide bond formation suggested that the 106-times higher reaction rate observed experimentally was attributable to entropy (i.e., the appropriate positioning of substrates at the PTC).6) Another study on the pH dependence of the PTC reaction found that it did not involve acid-base catalysis by ribosome residues.7) On the other hand, kinetic isotope effects on the reaction between aminoacyl-tRNAs and peptidyl-tRNAs were observed using carbon, nitrogen, and oxygen isotopes at the PTC, indicating that the reaction proceeds in a stepwise fashion with C–N bond formation as the rate-limiting step.8) A study on the kinetic solvent isotope effects of the peptidyl transfer reaction at the PTC in buffer solutions containing various concentrations of D2O showed that three protons participate in the transition state of peptide bond formation, with simultaneous transfer of protons of the NH2 group, the 2′-OH group of peptidyl-tRNA A76, and a water molecule.9)
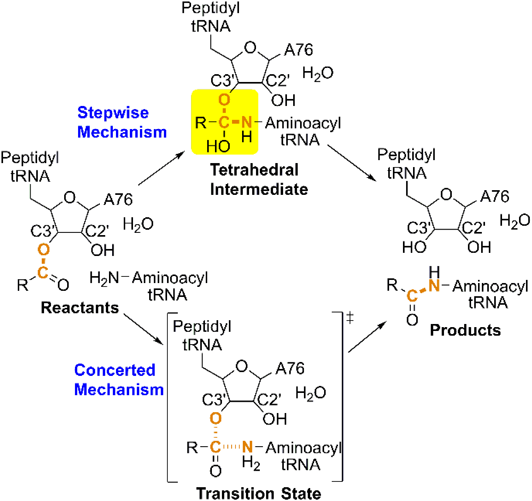
There are two competing theories on how peptide bond formation proceeds in the PTC (Fig. 1). The stepwise reaction involves C–N bond formation in the first step and C–O bond cleavage in the second step, with a tetrahedral intermediate.10,11) In contrast, C–N bond formation and C–O bond cleavage occur in one step through a single transition state in the concerted reaction. Proton transfer from the aminoacyl-tRNA NH2 group in the reactant to the 3′-OH group of peptidyl-tRNA A76 in the product is common to both of these mechanisms. The 2′-OH group of peptidyl-tRNA A76, as well as the water molecule trapped in the PTC, are presumably incorporated into the proton transfer route and facilitate proton transfer through the hydrogen-bonding network. Quantum chemical calculations of ribosome systems of various sizes have yielded concrete images of the mechanisms of peptide bond formation, including the geometry and energy of reaction intermediates and transition states. Quantum chemical studies on the mechanism of amide formation from corresponding esters or carboxylic acid in the gas phase suggested a stepwise mechanism through a tetrahedral intermediate.10–13) However, there is also evidence for a concerted mechanism.14–19) In a PTC model system comprising 76 atoms, calculated based on the B3LYP/6-311G** level of theory, C–N bond formation and C–O bond cleavage in the transition state occurred simultaneously with proton transfer in a cyclic hydrogen-bonding system that included the ester C=O group, NH2 group, and 2′-OH group of A76; furthermore, the water molecule trapped at the PTC participated in the hydrogen-bonding network and reduced the activation energy of peptide bond formation.19) The latter was supported by QM/MM free energy simulations.18)
Quantum chemical calculations have been used to study both stepwise and concerted mechanisms.20–31) In one study of systems containing 44–56 atoms of the PTC analyzed using the M06-2X/6-311 + G(d,p) level of theory, C–N bond formation and C–O bond cleavage were accompanied by proton transfer of the NH2 group, 2′-OH group of A76, and water molecules in the transition states (proton-shuttle mechanism); these authors showed that a stepwise reaction was favored over a concerted one and that the 2′-OH group of rRNA A2451 in the active site of the ribosome stabilized the transition states through hydrogen bonding.22)
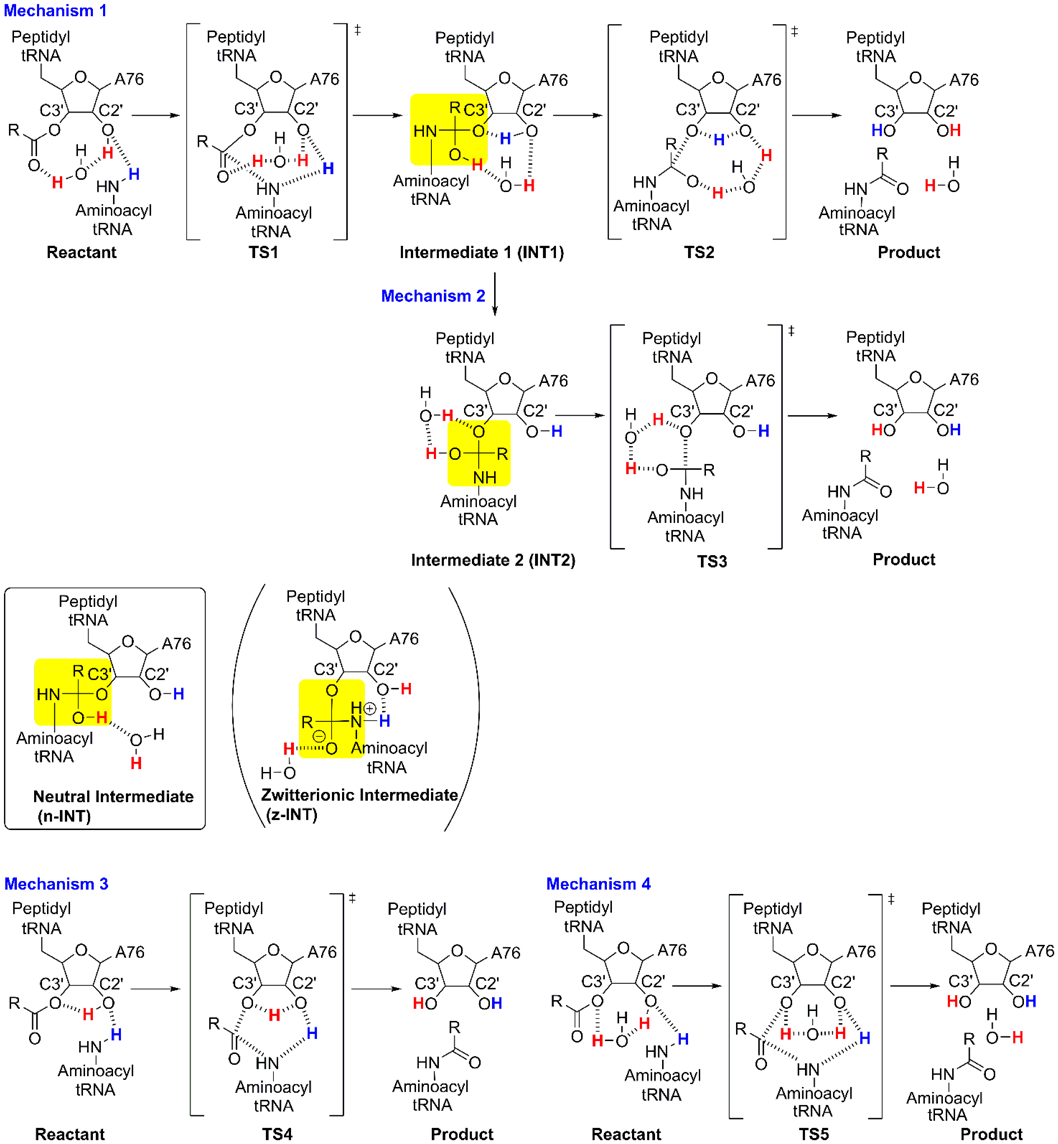
In a previous study, we demonstrated the stepwise mechanism of the peptidyl transfer reaction in a ribosome system consisting of 2354 atoms using the ONIOM method23) (Fig. 2, Mechanism 1). This approach can be used to quantitatively analyze the environmental effects of enzymatic reactions in large systems. We assigned the entire molecular system to a real system; the reaction center comprising 64 substrate atoms and the PTC were assigned to the model system for ONIOM(B3LYP/6-31G(d,p):Amber) calculations. The transition states for C–N bond formation (TS1) and C–O bond cleavage (TS2) were accompanied by the transfer of three protons from the NH2 group, the 2′-OH group of A76, and a water molecule. The calculated energy barriers (ΔE) for TS1 and TS2 were 24.3 and 51.9 kcal mol−1, respectively; the latter was too large to account for the experimentally determined energy barrier of 17 kcal mol−1.32)
Based on these studies, in the current study, we compared the stepwise and the concerted mechanisms of a larger ribosomal system than those used in previous studies. Furthermore, preliminary surveys in the current study showed that mechanism 2 has comparable activation energy to mechanism 1. From this series of studies, we decided to compare the four mechanisms shown in Fig. 2 using the ONIOM calculations. The calculations were carried out using the ONIOM(M06-2X/def2-TZVPP:Amber)//ONIOM(M06-2X/6-31G(d,p):Amber) method. The entire calculated system comprised 2354 atoms assigned to a real system, and the reaction center included 180 atoms that were assigned to the model system. The large model system allowed us to consider electrostatic and steric effects on intermediates and transition states of peptide bond formation proceeding in the PTC of the ribosome under steric constraints imposed by factors outside the system.
Results and Discussion
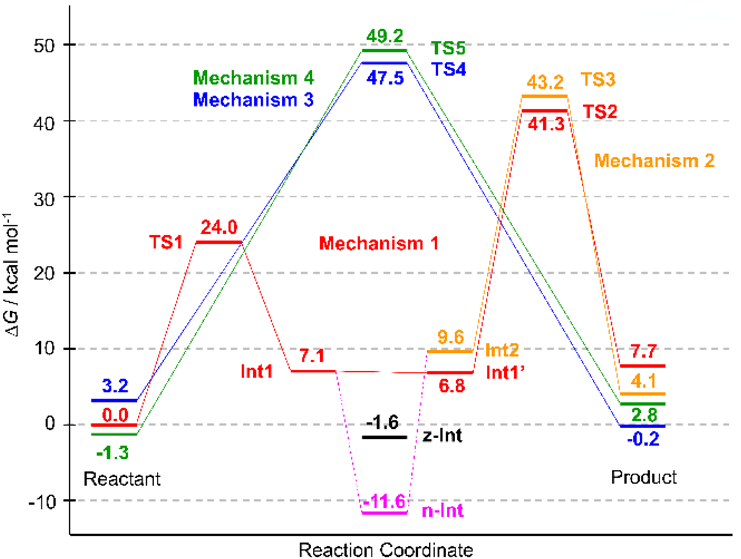
The four mechanisms for which calculations were performed are shown in Fig. 2. Mechanism 1 is a stepwise reaction through the neutral tetrahedral intermediate 1 (INT1) and its conformer (INT1′), both of which have almost identical energies and geometries (Supplementary Materials, Tables S1–S7, Figs. S4 and S5). The slight differences between INT1 and INT1′ indicate that these species are not identical and suggest that interconversion between them is possible.
The C–N bond of INT1 is formed by nucleophilic attack by the NH2 group of aminoacyl-tRNA on the carbonyl group through TS1. The C–O bond of INT1′ is cleaved by the 3′-O of peptidyl-tRNA A76 leaving through TS2. Although the role of the 2′-OH group of A76 of peptidyl-tRNA is debatable,33–37) we adopted a substrate-assisted catalysis model in which the 2′-OH group of A76 catalyzes peptide bond formation,37) since this is consistent with experimental data and calculated models from previous mechanistic studies.8,9,12–14,16–31,33,34)
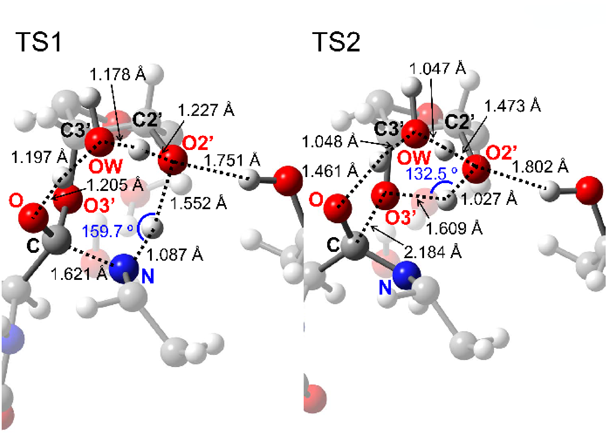
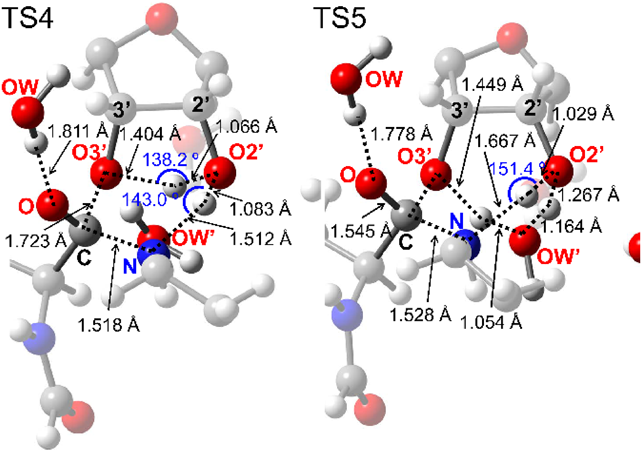
The optimized geometries of TS1 and TS2 are shown in Fig. 3. TS1 involved the sequential transfer of three protons (red and blue protons in Fig. 2) from the NH2 group to the ester C=O group via the A76 2′-OH group and the trapped water molecule. This transfer enhanced the nucleophilicity of the NH2 group and electrophilicity of the C=O group, which cooperatively promoted C–N bond formation of INT1. TS2 resulting in peptide bond formation involved a similar sequential transfer of three protons from the OH group of a tetrahedral intermediate center to the 3′-O of A76 via the 2′-OH group of A76 and the trapped water molecule. The protons neutralized the negative charge that developed on the A76 3′-O-leaving group, thereby accelerating C–O bond cleavage. The protons of the 2′-OH group of A76 and the water molecule (red protons in Fig. 2) moved to the water molecule and ester C=O group in the first step and returned to their initial positions in the second step. The shuttling of these protons facilitated peptide bond formation and proton transfer from the NH2 group to the 3′-O of A76 (blue proton in Fig. 2).
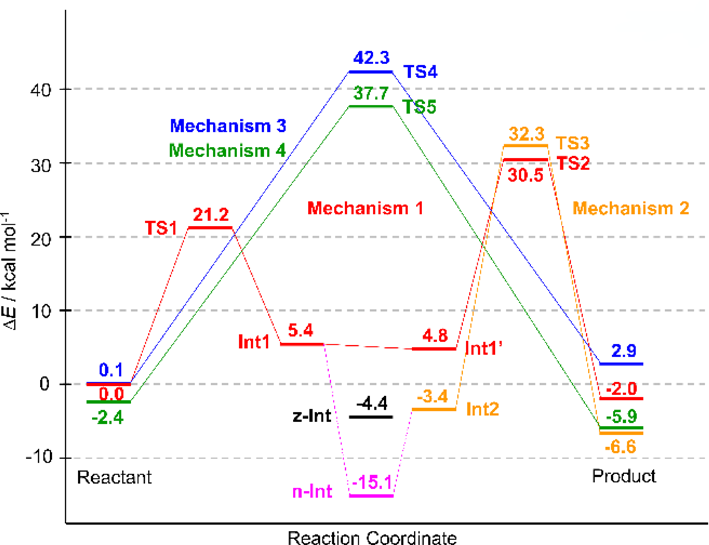
The calculated relative energies (ΔE) of the TS1 and TS2 model systems were 21.2 and 30.5 kcal mol−1, respectively (Fig. 4), which are lower than the values obtained in our previous study (24.3 and 51.9 kcal mol−1, respectively).23) This decreased energy barrier indicates that intermolecular interactions such as electrostatic and van der Waals interactions between substrates and the PTC environment stabilize the transition states, especially TS2. Although it is very difficult to quantitatively evaluate the effect of the PTC environment on the reaction mechanism in the present calculations, Figs. S14 and S15 show that the dipole direction of the 180-atom system is different from that of the 76-atom system for both TS1 and TS2. This difference can be attributed to the influence of polar residues such as phosphate groups and nucleobases in the 180-atom system.
Despite the large stabilization of TS2, the energy profile showed that TS2 has a higher barrier than TS1 (Fig. 4), indicating that C–O bond cleavage is the rate-limiting step. Although this is inconsistent with the results of kinetic studies showing that the formation of the tetrahedral intermediate is rate-limiting,8) a similar trend has been reported in theoretical calculations of stepwise amide or peptide bond formation.10–13,20–27,29–31)
According to Rangelov et al., the height of the energy barrier of peptide bond formation is related to the angular deviation of the proton transfer route (O2′-H-O3′ or N-H-O2′) from linearity in the transition state.12) Therefore, the larger angular deviation of ΔθO2′-H-O3′ = 47.5° at TS2 compared to that of ΔθN-H-O2′ = 20.3° at TS1 supports our conclusion that the rate-limiting step is TS2.
Calculations of natural atomic charges of TS1, TS1′, TS1″, TS2, TS2′, and TS2″ were carried out (Supplementary Materials, Tables S39–S41). TS1′ and TS2′ were made from TS1 and TS2 by removing the H2O molecule that interacts with O and H2′. TS1″ and TS2″ were made from TS1′ and TS2′ by replacing the 2′-OH group with an H atom. The calculated charges show that the O of the C=O group (O), 2′-O (O2′), and the O of the H2O molecule (Ow) in the catalytic site have large negative charges (−0.850, −0.891, and −1.080) in TS1, while the O of the tetrahedral intermediate (O), O2′, O3′, and Ow have large negative charges (−0.720, −0.832, −1.024, and −0.893) in TS2. Comparison of the charges of TS1, TS1′, and TS1″ shows that the hydrogen bonds in TS1 stabilize the negative charge of O, and facilitates the proton transfer. On the other hand, comparison of the charges of TS2, TS2′, and TS2″ shows that the negative charge generated on O3′ during the cleavage process of the ester bond is not sufficiently neutralized by the hydrogen-bonding network of TS2. In addition to the large angular deviation of the hydrogen bonds, this may be one of the reasons for the high energy of TS2.
The relative Gibbs energies (ΔG) of TS1 and TS2 were 24.0 and 41.3 kcal mol−1, respectively (Fig. 5). These values were obtained from the relative enthalpies (ΔH) of 19.1 and 37.0 kcal mol−1 and relative entropies (TΔS) of −4.9 and −4.3 kcal mol−1 of TS1 and TS2, respectively (Supplementary Materials, Table S8). The large ΔH of TS2 indicated that TS2 stabilization was insufficient for the entire calculated system.
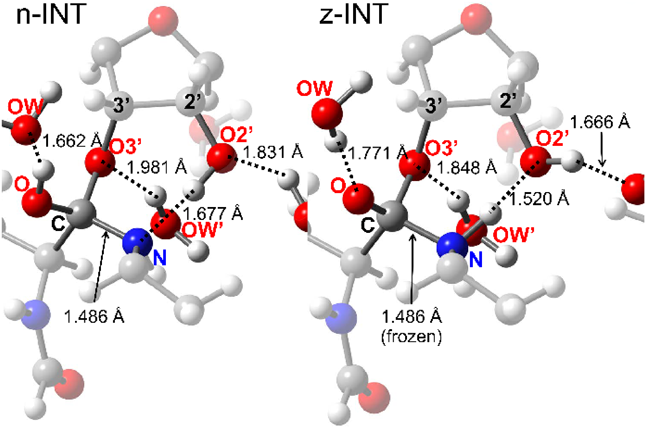
Mechanism 2 involves a stepwise transition through INT2 and TS3 (Fig. 2). The conformational change of INT1 yields INT2, leading to product formation through TS3 (Fig. 6). A neutral tetrahedral intermediate (n-INT) was discovered by searching along the route from INT1 to INT2 (Fig. 7). The optimized structure indicated that n-INT was stabilized in the PTC environment through multiple hydrogen bonds.
A study based on the X-ray crystal structures of the Haloarcula marismortui 50S ribosome suggested the existence of a zwitterionic intermediate that breaks down through deprotonation of ammonium ion via the 2′-OH group of A76 of aminoacyl-tRNA to yield a peptide bond.33) An alternative ionic intermediate was suggested based on the X-ray crystal structures of the complexes before and after peptide bond formation on the Thermus thermophilus 70S ribosome.38) The negative charge on the oxygen atom of the tetrahedral intermediate was stabilized by hydrogen bonding with the trapped water molecule. The positive charge was located on the hydronium ion, which may be stabilized by interaction with the 2′-OH group of A2451, the phosphate group of A76 of aminoacyl-tRNA, and the amino group of the L27 peptide. The intermediate breaks down through the proton wire that consists of the trapped water molecule, the 2′-OH group of A2451, the 2′-OH group of A76 of peptidyl-tRNA, and the hydronium ion. Although kinetic experiments showed that the amino group of the L27 peptide did not affect the rate of peptide bond formation,39) the M06-2X/6-31 + G(d) calculation of the cluster model showed that the proton wire mechanism, in which the phosphate group of A76 of aminoacyl-tRNA interacts with the hydronium ion, is feasible.30) Calculation of the stepwise mechanism of peptide bond formation including such a zwitterionic intermediate using the M06-2X/6-31G(d,p) level of theory showed that the energy barrier was lower than that for the concerted mechanism.31) A similar zwitterionic species was characterized as a transient entity between the reactant complex and the transition state of peptide bond formation using QM/MM methods.24,28)
We tried to locate such a zwitterionic intermediate using ONIOM calculations and obtained an optimized geometry (Figs. 2 and 7, z-INT) when the C–N bond was frozen at 1.486 Å, which is the same length as the corresponding C–N bond in n-INT (Fig. 7). However, z-INT was not obtained as a stable species in our calculation conditions. The energy of the obtained z-INT was 10.0 kcal mol−1 higher than that of n-INT, and all attempts to optimize z-INT without restricting C–N bond length were unsuccessful.
The geometries of INT1 and n-INT are compared in Figs. S8–S10 (Supplementary Materials). INT1 has cyclic hydrogen bonding that consists of 3′-OH, H2O, 2′-OH, and an amino group. The cyclic hydrogen-bonding system was distorted, and the hydrogen-bonding angle at the 3′-OH and H2O was 95.99°. On the other hand, the corresponding hydrogen-bonding angle of n-INT was 111.36°, which is an undistorted and almost tetrahedral bond angle. In addition, Figs. S10 and S11 (Supplementary Materials) show that the position of the water molecule is similar in n-INT and the X-ray structure of 1vqp. Therefore, the stability of n-INT seems to come from the relief of the strain energy of the cyclic hydrogen-bonding system of INT1.
Transformation of INT2 yielded the product through cleavage of the C–O bond of the A76 3′-ester group, which was accompanied by proton transfer in TS3 (Figs. 2, 6). The proton of the OH group of the INT2 tetrahedral intermediate was transferred to the trapped water molecule, whose proton was transferred to the A76 3′-O. This neutralized the charges that developed on the latter, which in turn facilitated C–O bond cleavage. The ΔE of 32.3 kcal mol−1 and ΔG of 43.2 kcal mol−1 in TS3 were obtained by ONIOM calculations (Figs. 4, 5). The barrier height of TS3 was comparable to that of TS2, which was confirmed by the similar angular deviations of TS3 (ΔθO-H-O3′ = 40.0°) and TS2 (ΔθO2′-H-O3′ = 47.5°).
Mechanisms 3 and 4 are concerted mechanisms that proceed through TS4 and TS5 (Figs. 2, 8). C–N bond formation and C–O bond cleavage in TS4 were accompanied by the transfer of two protons—i.e., from the NH2 group to the A76 2′-OH group and from the 2′-OH group to the 3′-O of A76. C–N bond formation and C–O bond cleavage at TS5 were accompanied by the transfer of three protons—i.e., from NH2 to A76 2′-OH, from A76 2′-OH to the trapped water molecule, and from the water molecule to A76 3′-O. Although the cooperative proton transfer at TS4 and TS5 facilitated C–N bond formation and C–O bond cleavage, the energy barriers of these reactions were higher than those of TS1–TS3 (Figs. 3, 6), indicating that the concerted mechanisms are less favorable than the stepwise ones.
Our calculations have two limitations. First, the obtained activation energy of the rate-determining step was much higher than the experimentally obtained value.32) Our previous study was carried out using a small model system containing 76 atoms and adopted the B3LYP/6-31G(d,p) method for the optimization of the model system.23) In this study, we used a larger model system containing 180 atoms and adopted the M06-2X/6-31G(d,p) method for the optimization of the model system. The comparison of these calculations suggested that the energy of TS2 was stabilized by short-range intermolecular interactions. However, the calculation conditions in this study did not include long-range interactions such as the electrostatic effects of the surroundings of the model system, which include phosphate anions and Mg2+, Na+, and K+ cations, in the reaction center. Therefore, our calculations yielded qualitative results, even though the calculation conditions were improved in comparison to those of previous studies.
Second, the potential energy surface including that of n-INT was not obtained in our study. The geometry comparison of n-INT, INT1, and the reaction center of 1vqp showed that n-INT was less distorted than INT1 and that the geometry of n-INT was reasonable based on the X-ray structure. Although we could not clarify the reaction path through n-INT, energy profiles in Figs. 4 and 5 show that such reaction path would have very high activation energy at the step from n-INT to TS2 or TS3. To examine whether n-INT is involved in the peptide bond formation, we carried out model calculations of the reaction pathways from INT1 to INT1′ and from INT1 to n-INT by the nudged elastic band method40,41) using Reaction plus Pro.42) As a result, the process from INT1 to INT1′ was found to be a small conformational change with a low energy barrier (Supplementary Materials, Figs. S6 and S7, Table S1). On the other hand, the process from INT1 to n-INT consists of dissociations and formations of hydrogen-bonding of the H2O molecule in the active site, which has a large energy barrier (Supplementary Materials, Figs. S12 and S13, Table S11). Although these results cannot be directly compared with the ONIOM calculations, they seem to suggest that the reaction pathway from INT1 to INT1′ is kinetically preferable to that from INT1 to n-INT.
Conclusion
In conclusion, we compared four mechanisms of peptide bond formation on the ribosome based on ONIOM calculations of the 2354-atom system and found that 1) the transition states of the stepwise mechanisms TS1, TS2, and TS3 had lower energies compared with those of the concerted ones TS4 and TS5; and 2) n-INT exists as a stable species in the stepwise reaction, while z-INT was not detected by unrestricted optimization.
The stepwise mechanisms that we obtained proceed via a neutral intermediate and are consistent with traditional calculated mechanisms for small model systems.10–13) The optimized geometries showed that the development of charges on the NH2 group of aminoacyl-tRNA, the ester C=O group, and the 3′-O of the peptidyl-tRNA A76 moiety in transition states are neutralized by simultaneous proton transfer through the hydrogen-bonding network, which includes water molecules in the PTC environment. Moreover, our model involves three proton transfers in the rate-determining step, which is consistent with the proton inventory experiment reported by Kuhlenkoetter et al.9) These results indicate that the stepwise proton-shuttle mechanism seems plausible in the PTC environment of the ribosome.
However, if n-INT is the intermediate of the stepwise mechanism, the energy barrier from n-INT to TS2 or TS3 would be very high, and thus these processes do not become the favored route. Therefore, clarifying the involvement of n-INT in the reaction path would be an important step to understanding the mechanism of peptide bond formation on the ribosome.
Experimental
All quantum chemical calculations were performed using Gaussian09 D.01.43) Input files for calculations were prepared based on the X-ray crystallographic data of the 50S subunit of the ribosome of H. marismortui obtained from the Protein Data Bank (PDB ID: 1vqp).33) A region within 20 Å from the center of the PTC was extracted from the X-ray data (Fig. S1). The structure of the extracted region was modified for the calculations of the mechanisms shown in Fig. 2. The detailed procedure of modification is the same as in our previous study23) and is described in the Supplementary Materials.
The entire calculated system was divided into the real and model systems and was calculated using the ONIOM method44,45) (Fig. 9). The real system contained 2354 atoms and was calculated using the molecular mechanics method using the Amber force field.46) The model system contained 180 atoms (Fig. 10) and was calculated using the M06-2X functional quantum chemical method.47,48) The model system included the substrates, the functional groups that directly interact with the substrates through hydrogen bonds, and the surroundings that indirectly affect the substrates through steric and electrostatic effects.
Optimization of the entire system was carried out using the ONIOM(M06-2X/6-31G(d,p):Amber) method. Similarly, the single-point energy calculation was performed using the ONIOM(M06-2X/def2-TZVPP:Amber) method, with a density fitting basis set49) that describes the model system quantum-mechanically. Electronic embedding was not used for geometry optimization and single-point energy calculation.
All the stationary points obtained by geometry optimization were confirmed as saddle points or energy minima by vibrational frequency analysis. Gibbs energies were obtained by the addition of enthalpy and entropy correction from the frequency calculation of the energy of stationary points on the potential energy surface.
Acknowledgment
This work was supported by a Japan Society for the Promotion of Science KAKENHI Grant (no. 25330343).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
References
- 1) Rheinberger H.-J., “Protein Synthesis and Ribosome Structure,” ed. by Neirhous K. H., Wilson D. N., Wiley-VCH, Weinheim, 2004, pp. 1–52.
- 2) Nierhous K. H., “Protein Synthesis and Ribosome Structure,” ed. by Neirhous K. H., Wilson D. N., Wiley-VCH, Weinheim, 2004, pp. 323–366.
- 3) Liljas A., Ehrenberg M., “Structural Aspects of Protein Synthesis,” 2nd ed., World Scientific Publishing Co., Pte. Ltd., Singapore, 2013, pp. 261–302.
- 4) Borman S., Chem. Eng. News, 82, 29–30 (2004).
- 5) Ban N., Nissen P., Hansen J. P., Moore B., Steitz T. A., Science, 289, 905–920 (2000).
- 6) Sievers A., Beringer M., Rodnina M. V., Wolfenden R., Proc. Natl. Acad. Sci. U.S.A., 101, 7897–7901 (2004).
- 7) Bieling P., Beringer M., Adio S., Rodnina M. V., Nat. Struct. Mol. Biol., 13, 423–428 (2006).
- 8) Hiller D. A., Singh V., Zhong M., Strobel S. A., Nature (London), 476, 236–239 (2011).
- 9) Kuhlenkoetter S., Wintermeyer W., Rodnina M. V., Nature (London), 476, 351–354 (2011).
- 10) Oie T., Loew G. H., Burt S. K., MacElroy R. D., J. Am. Chem. Soc., 105, 2221–2227 (1983).
- 11) Ilieva S., Galabov B., Musaev D. G., Morokuma K., Schaefer H. F. III, J. Org. Chem., 68, 1496–1502 (2003).
- 12) Rangelov M. A., Vayssilov G. N., Yomtova V. M., Petkov D. D., J. Am. Chem. Soc., 128, 4964–4965 (2006).
- 13) Rangelov M. A., Petrova G. P., Yomtova V. M., Vayssilov G. N., J. Org. Chem., 75, 6782–6792 (2010).
- 14) Sharma P. K., Xiang Y., Kato M., Warshel A., Biochemistry, 44, 11307–11314 (2005).
- 15) Gindulyte A., Bashan A., Agmon I., Massa L., Yonath A., Karle J., Proc. Natl. Acad. Sci. U.S.A., 103, 13327–13332 (2006).
- 16) Kästner J., Sherwood P., Mol. Phys., 108, 293–306 (2010).
- 17) Monajemi H., Zain S. M., Abdullah W. A. T. W., Comput. Theor. Chem., 976, 148–152 (2011).
- 18) Xu J., Zhang J. Z. H., Xiang Y., J. Am. Chem. Soc., 134, 16424–16429 (2012).
- 19) Wallin G., Åqvist J., Proc. Natl. Acad. Sci. U.S.A., 107, 1888–1893 (2010).
- 20) Trobro S., Åqvist J., Proc. Natl. Acad. Sci. U.S.A., 102, 12395–12400 (2005).
- 21) Wang Q., Gao J., Liu Y., Liu C., Chem. Phys. Lett., 501, 113–117 (2010).
- 22) Acosta-Silva C., Bertran J., Branchadell V., Oliva A., J. Am. Chem. Soc., 134, 5817–5831 (2012).
- 23) Fukushima K., Iwahashi H., Nishikimi M., Bull. Chem. Soc. Jpn., 85, 1093–1101 (2012).
- 24) Świderek K., Tuñón I., Martí S., Moliner V., Bertrán J., Chem. Commun., 48, 11253–11255 (2012).
- 25) Świderek K., Tuñón I., Martí S., Moliner V., Bertrán J., J. Am. Chem. Soc., 135, 8708–8719 (2013).
- 26) Byun J. B., Kang Y. K., Phys. Chem. Chem. Phys., 15, 14931–14935 (2013).
- 27) Monajemi H., Zain S. M., Abdullah W. A. T. W., RSC Adv., 5, 25489–25503 (2015).
- 28) Świderek K., Marti S., Tuñón I., Moliner V., Bertran J., J. Am. Chem. Soc., 137, 12024–12034 (2015).
- 29) Zhang X., Jiang Y., Mao Q., Tan H., Li X., Chen G., Jia Z., Molecules, 22, 571 (2017).
- 30) Acosta-Silva C., Bertran J., Branchadell V., Oliva A., Theor. Chem. Acc., 136, 49 (2017).
- 31) Kazemi M., Sočan J., Himo F., Åqvist J., Nucleic Acids Res., 46, 5345–5354 (2018).
- 32) Johansson M., Bouakaz E., Lovmar M., Ehrenberg M., Mol. Cell, 30, 589–598 (2008).
- 33) Schmeing T. M., Huang K. S., Kitchen D. E., Strobel S. A., Steitz T. A., Mol. Cell, 20, 437–448 (2005).
- 34) Weinger J. S., Parnell K. M., Dorner S., Green R., Strobel S. A., Nat. Struct. Mol. Biol., 11, 1101–1106 (2004).
- 35) Koch M., Huang Y., Sprinzl M., Angew. Chem. Int. Ed., 47, 7242–7245 (2008).
- 36) Huang Y., Sprinzl M., Angew. Chem. Int. Ed., 50, 7287–7289 (2011).
- 37) Zaher H. S., Shaw J. J., Strobel S. A., Green R., EMBO J., 30, 2445–2453 (2011).
- 38) Polikanov Y. S., Steitz T. A., Innis C. A., Nat. Struct. Mol. Biol., 21, 787–793 (2014).
- 39) Maracci C., Wohlgemuth I., Rodnina M. V., RNA, 21, 2047–2052 (2015).
- 40) Jónsson H., Mills G., Jacobsen K. W., “Classical and Quantum Dynamics in Condensed Phase Simulations,” ed. by Berne B. J., Ciccotti G., Coker D. F., World Scientific, 1998, pp. 385–404.
- 41) Henkelman G., Jónsson H., J. Chem. Phys., 113, 9978–9985 (2000).
- 42) HPC Systems Inc., “Software to optimize reaction paths along the user’s expected ones.”: ‹http://www.hpc.co.jp/chem/software/react1.html›, cited 29 June, 2021.
- 43) Frisch M. J., Trucks G. W., Schlegel H. B., et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT (2016).
- 44) Svensson M., Humbel S., Froese R. D. J., Matsubara T., Sieber S., Morokuma K., J. Phys. Chem., 100, 19357–19363 (1996).
- 45) Chung L. W., Sameera W. M. C., Ramozzi R., Page A. J., Hatanaka M., Petrova G. P., Harris T. V., Li X., Ke Z., Liu F., Li H.-B., Ding L., Morokuma K., Chem. Rev., 115, 5678–5796 (2015).
- 46) Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M. Jr., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A., J. Am. Chem. Soc., 117, 5179–5197 (1995).
- 47) Zhao Y., Truhlar D. G., Theor. Chem. Acc., 120, 215–241 (2008).
- 48) Zhao Y., Truhlar D. G., Acc. Chem. Res., 41, 157–167 (2008).
- 49) Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys., 7, 3297–3305 (2005).