Abstract
The particles of phenytoin (Phe), a poorly water-soluble model drug, were bead-milled alone or co-milled with a hydrophilic waxy additive using an ultra cryo-milling technique in liquid nitrogen (LN2) to improve its dissolution properties. However, the micronized drug particles adhered and aggregated, resulting in poor handling in manufacturing processes such as blending or tableting. To improve the dissolution profile and powder properties of the drug simultaneously, the milled products were secondarily processed together with larger spherical particles by mechanical powder processing. These secondary products were composite particles with a core-shell structure, with fine drug particles adhered and deposited on the core, based on order mixing theory. As a core, three types/sizes of spherical pharmaceutical excipient particles were applied. The resultant composite particles produced much faster release profiles than just milled or co-milled mixtures. In addition, the composite particles showed good micromeritic properties depending on the size of the core particles. These results indicate that the ultra cryo-milling and subsequent dry composite mixing is a potential approach for developing drug particles with improved dissolution.
Introduction
The oral solid dosage is the most popular form for drugs that must be taken daily, because of its convenience and functionality. After administration, the drug is eventually absorbed from the gastrointestinal membrane in a solution state through the processes of disintegration (of tablets, capsules, or granules), particle dispersion, and dissolution in the gastrointestinal tract. The dissolution of drug from the dosage form is the rate-limiting step in the drug absorption process for many pharmaceutical products, especially poorly water-soluble drugs. New hydrophobic and less water-soluble pharmaceutical candidates have been developed as a result of progress in highly sensitive in vitro affinity assays using sophisticated high-throughput screening equipment.1) Many of these poorly water-soluble drugs are categorized as biopharmaceutics classification system (BCS) class II for which their oral absorption is limited by their extremely poor solubility in the gastrointestinal tract.2) Therefore, improving dissolution is a critical issue for developing oral solid dosage forms. Traditional approaches, such as pH adjustment and salt formation, remain useful to improve dissolution, however, particle size reduction is also a good choice for developing drug products due to their wide applicability to many drug substances.3) Reducing particle size and increasing the surface area of drug substances can improve dissolution rates of drugs, according to the Noyes–Whitney equation.4) In general, wet milling in the aqueous phase often is used to obtain submicron-sized particles, but the resultant milled suspension has to be dried to develop tablets and capsules, which are the major solid dosage forms. However, dry milling can reduce particle size only to 2–3 µm, because of secondary co-aggregation occurring between particles as a result of van del Waals’ attractive forces.5) In addition, changes in the crystal characteristics (crystalline form, crystallinity) of the drug can be a problem due to the generation of heat by friction and shearing force between particles and milling media.
Ultra cryo-milling, in which drug particles are suspended in liquid nitrogen (LN2) and pulverized by small hard balls (beads) was developed and reported to improve the dissolution behavior of poorly water-soluble drugs.6) In this technique, LN2 was used as a dispersion solvent and the drug particles were effectively micronized by colliding with zirconia beads based on the brittle fracture generated under cryogenic temperature. This technique could also prevent the chemical and physicochemical degradation of drugs caused by the heat generated from pulverization. In addition, LN2 has the potential to be a good physical barrier against secondary co-aggregation during the milling process because it can penetrate into void spaces and micropores between and inside the particles due to its inherent low surface tension (8.85 mN/m at −196 °C, around one-eighth that of water) and low viscosity (0.158 mPa·s at −196 °C, around one-sixth that of water).7,8) Thus, ultra cryo-milling could achieve micronization of the particles down to the submicron size. In addition, LN2 is industrially available at a reasonable price and is inert, nonflammable, and nontoxic. Furthermore, since it is a very low-temperature liquid with a boiling point of −196 °C and spontaneously evaporates at ambient temperature and atmospheric pressure, the ultra cryo-milling is a general wet milling method that does not require a heating process to dry the milled powder because the LN2 is vaporized spontaneously. Thus, ultra cryo-milling is a hybrid method that has the advantages of both wet milling and dry milling.
However, the resulting fine-particulate products obtained through the ultra cryo-milling process are likely to aggregate and possess poor powder handling properties (low flowability and packability), which can be troublesome in manufacturing operations such as blending and tableting. Although a formulation technique to produce fine particles of 2–3 µm or less without aggregation has not yet been established, mechanical powder processing is a promising and attractive technology that offers easy handling.9) This technology involves the formation of composite particles with multi-components by providing powders through powerful shearing, impacting, and compressive forces using a high-shear processor. Such technology is also known as mechanofusion or hybridization. This process and its products are called “ordered mixing” and “ordered mixture (OM),” respectively, due to the well-regulated mixing style. Mechanical stress promotes the breakage of aggregated fine particles and the mixing of different materials, and can be used for dry particle coating and the precision mixing of nanoparticles. By forcibly transporting the powder through a gap of approx. 1 mm and applying intensive shear force between the aggregated fine particles and the core particles, the conglomerated fine particles are effectively de-aggregated and fixed uniformly onto the surface of core particles. Dry particle coating using a mechanical powder processor includes the arrangement and fixation of small guest particles onto large host particles based on an ordered mixing principle under high mechanical force. This process can also modify the surface by surrounding core particles by fine particles several microns in diameter in a solvent-less process. In the pharmaceutical industry, mechanical powder coating is utilized to improve flowability of cohesive powders, dispersibility of inhalational powders, and solubility of poorly water-soluble drugs.10)
In the present study, the poorly water-soluble model drug, phenytoin, was milled alone or co-milled with a water-soluble excipient using the ultra cryo-milling technique. Subsequently, the cryo-milled drug particles were mechanically processed with the core particles through the mechanical powder processing process, also called the dry composite mixing process. Spherical composite particles with an OM structure were prepared in two steps, during which ultra cryo-milled drug particles were deposited on the surface of core particles. Three types of spherical core particles with different diameters (starch, sugar alcohol, cellulose), which were over 10-fold larger than the drug particles, were used to prepare the composite particles. The main aim of the present study was to improve both the micromeritic and dissolution properties of micronized drug particles by preparing OM composite particles (OMs). In addition, a novel milling and dry processing technique was developed to produce spherical drug particles for fine particle coating to mask the bitter taste of the drug. This report describes the sequential technique through ultra cryo-milling and mechanical powder processing to produce spherical drug particles applicable to research and production activities in the pharmaceutical industry.
Experimental
Materials and MethodsPhenytoin was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Polyethylene glycol 6000 was provided by Sanyo Chemical Industries, Ltd. (Kyoto, Japan). Corn starch particles, spherical granules of D-mannitol, and microcrystalline cellulose were used as core particles in the dry composite mixing process. Japanese Pharmacopoeia (JP) grade corn starch (corn starch white) was provided by Japan Corn Starch Co., Ltd. (Tokyo, Japan). D-Mannitol spheres (JP grade; Nonpareil-108) was provided by Freund Corporation (Tokyo, Japan). Microcrystalline cellulose spheres [Japanese pharmaceutical excipient (JPE) grade; Celphere CP-203] was provided by Asahi Kasei Corporation (Tokyo, Japan). Phenytoin, polyethylene glycol 6000, corn starch particles, D-mannitol spheres, and microcrystalline cellulose spheres are referred to as Phe, PEG, CS, NP, and CP, respectively, in this report. LN2 for NMR was used (NS-100, Iwatani Industrial Gases Co., Ltd., Tokyo, Japan). All other chemicals and solvents were of analytical reagent grade, and deionized-distilled water was used throughout the study.
Manufacturing InstrumentsThe batch-type wet milling machine (RMB-04, Aimex Co., Ltd., Tokyo, Japan), which is generally used in wet-milling with beads in an aqueous solution, was improved to execute ultra cryo-milling in LN2. A schematic diagram of the equipment was published in a previous report.6) In this study, the vessel with a 400-mL capacity and its rotation shaft and disks were newly made of zirconia (zirconium oxide) while maintaining their original shape and size. Hard small zirconia balls, called “beads,” 1- and 0.6-mm in diameter (YTZ-1, -0.6), were purchased from Nikkato Co., Ltd. (Osaka, Japan) as milling media.
A mechanical powder processor (Nobilta-Mini, Hosokawa Micron Co., Ltd., Osaka, Japan) based on a mechanofusion principle was used for mechanical powder processing. This machine was composed mainly of a cylindrical vessel, rotator equipped with 8 blades, and a motor. The vessel and rotator were vertically positioned to allow sample powder to travel downward on the inner round wall of the vessel. Thus, the sample powder experienced powerful shearing forces while passing through the gap between the wall of vessel and the rotator blades, which was set to 1 mm in the present study. A schematic diagram of this machine was published previously.11)
Preparation of Cryo-Milled ParticlesPhe was milled alone using the ultra cryo-milling technique. The Phe was also co-milled with PEG, a water-soluble polymer, as a dispersion promotor in water. The weight ratio of Phe : PEG was set to 1 : 1. The formulated amounts of sample and zirconia beads (Table 1) were placed in a 400-mL zirconia vessel, followed by addition of 200 mL LN2 to disperse and cool the sample. When the mixture of Phe and PEG was milled, the bead size and volume were changed from the standard (0.6 mm, 100 mL) to large one (1.0 mm, 50 mL) to avoid the severe adhesion of PEG powders to beads. The powder was dispersed uniformly and agitated during a pre-cooling process (300 rpm, 5 min) and then milled at 1600 rpm for 15 min. The LN2 was added periodically as it evaporated to maintain the volume of the dispersing liquid throughout the milling process (approximately 10 L of LN2 was used for each trial). After milling, the drug slurry was separated from the beads by passing through a 212-µm sieve. Milled Phe and co-milled Phe with PEG are referred to as M-Phe and M-(Phe + PEG), respectively.
Table 1. Formulation of Ultra Cryo-Milling
| Symbol | Loading (g) | Milling mediaa) |
|---|
| Intact Phe | Intact PEG | Size (mm) | Volume (mL) |
|---|
| M-Phe | 15.00 | — | 0.6 | 100 |
| M-(Phe + PEG) | 7.50 | 7.50 | 1.0 | 50 |
The cryo-milled M-Phe and M-(Phe + PEG) were secondarily treated with the core particles using a mechanical powder processor. The spherical compounds used as core particles were CS particles, NP spheres, and CP spheres. Average particle sizes of CS, NP, and CP were approximately 20, 100, and 270 µm, respectively. The loading amount was adjusted to produce a weight ratio of core particle to drug was of 9 : 1 (Table 2), and the total bulk volume of the powders into the vessel was 30 mL for the CS series and 20 mL for the NP and CP series to avoid overloading the electricity consumption of the motor. For the PEG formulation, the weight ratio of Core : Phe : PEG was 9 : 1 : 1 theoretically. All formulations were named CS9 : 1 : 0 or CS9 : 1 : 1, etc., based on the core and weight ratio of Core : Phe : PEG. Apart from the 9 : 1 weight ratio series, the composition of core and co-milled mixtures was changed to confirm the coating efficiency of the core particles only for NP. The amount of the core and co-milled mixture was changed so that the weight ratio of core to milled products was 9 : 2, 6 : 2, and 3 : 2 (abbreviated NP9 : 1 : 1, NP6 : 1 : 1, NP3 : 1 : 1); and 20 mL of the total powder was loaded to the processor (as shown in Table 3). The sample powder was placed into the vessel, and a cover equipped with a temperature indicator was attached. After the vessel was set vertically, rotation was started. The rotation speed was increased gradually up to 3000 rpm for 1.5 min to mix and distribute the sample powder throughout the vessel. Rotation was continued at the fixed speed of 3000 rpm for 10 min while monitoring the temperature and the electric power consumption in the vessel. All runs were conducted while circulating water at 20 °C in a jacket around the vessel to maintain a temperature below 30 °C. After processing, the product was transferred into the receiver by turning the vessel upside-down. The aggregate adhering to the vessel wall and blades was recovered by manual scraping.
Table 2. Formulation for the Dry Composite Mixing Process with Various Core Particles When Using a Mechanical Powder Processor
| Series (Core) | Symbol (Core : Phe : PEGa)) | Loading (g) | Bulk volume (mL) |
|---|
| Core | M-Phe | M-(Phe + PEG) |
|---|
| CS | CS9 : 1 : 0 | 11.70 | 1.30 | — | 30 |
| CS9 : 1 : 1 | 11.70 | — | 2.60 | 30 |
| NP | NP9 : 1 : 0 | 11.25 | 1.25 | — | 20 |
| NP9 : 1 : 1 | 9.45 | – | 2.10 | 20 |
| CP | CP9 : 1 : 0 | 13.16 | 1.46 | — | 20 |
| CP9 : 1 : 1 | 11.29 | — | 2.51 | 20 |
a) Core : Phe : PEG = core particles (CS or NP or CP) : phenytoin : polyethylene glycol 6000. Processing time: 10 min, rotor speed: 3000 rpm, clearance between vessel wall and rotor: 1 mm.
Table 3. Formulation for the Dry Composite Mixing Process with Various Contents (Core: Milled Product) When Using a Mechanical Powder Processor (Only the NP Series)
| Series (Core) | Symbol (Core : Phe : PEGa)) | Loading (g) | Bulk volume (mL) |
|---|
| Core | M-Phe | M-(Phe + PEG) |
|---|
| NP | NP9 : 1 : 1 | 8.10 | — | 1.80 | 20 |
| NP6 : 1 : 1 | 6.60 | — | 2.20 | 20 |
| NP3 : 1 : 1 | 4.20 | — | 2.80 | 20 |
a) Core : Phe : PEG = NP : phenytoin : polyethylene glycol 6000. Processing time: 10 min, rotor speed: 3000 rpm, clearance between vessel wall and rotor: 1 mm.
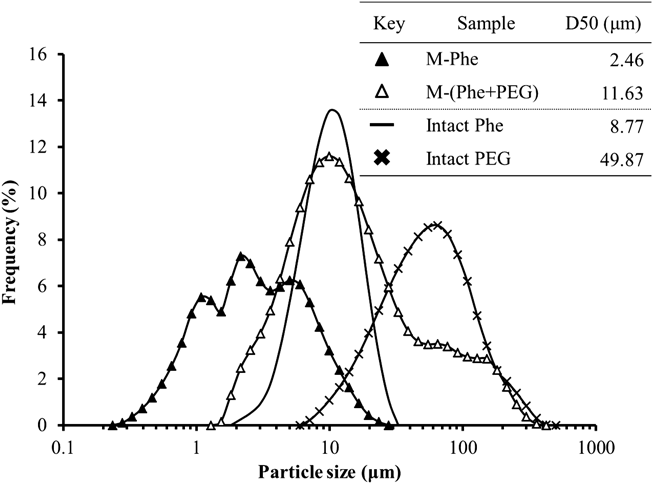
The morphology of the intact particles, cryo-milled particles, and mechanically processed particles was observed with a scanning electron microscope (SEM) (JSM-6060, Jeol Ltd., Tokyo, Japan) after coating using a platinum sputtering instrument (JFC-1600, Jeol Ltd.). The PSDs were determined using a laser diffraction scattering method with a diffractometer (LMS-30, Seishin Enterprise Co., Ltd., Tokyo, Japan). The dried particles were dispersed into dry air at a fixed air pressure of 0.4 MPa (dry conditions). The data obtained were plotted in a frequency volume distribution graph as a function of logarithmic particle size. Diameters at 50% of the cumulative volume distribution, designated D50 or volume median diameter, represented mean size.
True DensityThe particle densities of Phe and NP were measured using a helium pycnometer (Ultrapycnometer 1000, Quantachrome Corporation, FL, U.S.A.).
Drug Content and Recovery in ProductsThe drug content in the cryo-milled and the mechanically processed particles was measured to evaluate drug recovery in the products. Approximately 20 mg of the powder was weighed accurately and dissolved in 200 mL of aqueous 50% methanol solution, followed by filtration through a hydrophilic polytetrafluoroethylene (PTFE) membrane filter with a 0.20-µm pore size (Dismic-13HP, Advantec Toyo, Tokyo, Japan). The filtrate was diluted in methanol to the appropriate concentration and the quantity of Phe dissolved was assayed spectrophotometrically at 258 nm by HPLC (LC-10, Shimadzu Co., Ltd., Kyoto, Japan) equipped with a reverse-phase column (Inertsil ODS-3, 5 µm, 4.6 × 150 mm, GL Sciences, Tokyo, Japan). The Phe peak eluted at approximately 5 min when running mobile phase (acetonitrile: 10 mM Na2HPO4 solution, 11 : 9 v/v) at a flow rate of 0.8 mL/min. The percentage of actual drug amount to theoretical amount (mg/100 mg of product) was defined as the Phe recovery (%) using Eq. (1).
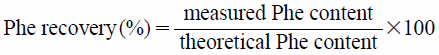 | (1) |
To evaluate the amount of drug that formed the OM, i.e., strongly fixed onto the surface of core particles, the mechanical processed products were separated through a sieve using an air-jet sieving apparatus (Spin Air Sieve SAM-75, Seishin Enterprise Co., Ltd.). A spoonful of powdered product was placed on the sieve with 45-µm openings and vacuumed with cyclone-type air flow at 1.5 kPa of air pressure to remove the free drug particles that were not fixed on the surface of the core particles. This sieving operation was repeated until enough sample was obtained for the assay. However, this operation was not suitable for the products in the CS series because their size was too small to separate free Phe particles through the sieve. The sample remaining on the sieve was also analyzed by HPLC using the same process as above. The retention ratio (%) was defined according to Eq. (2) to evaluate the entrapment efficiency of the drug on the surface of the composite particles:
 | (2) |
To evaluate the coating state more thoroughly, the number of coating layers of Phe was calculated arithmetically for only the NP-based composite particles. Considering particle size and particle density of NP and Phe particles, the Phe content was determined to be 5.00 mg/100 mg of OM when coated with a single-particle layer. Therefore, the number of coating layers was calculated from Eq. (3):
 | (3) |
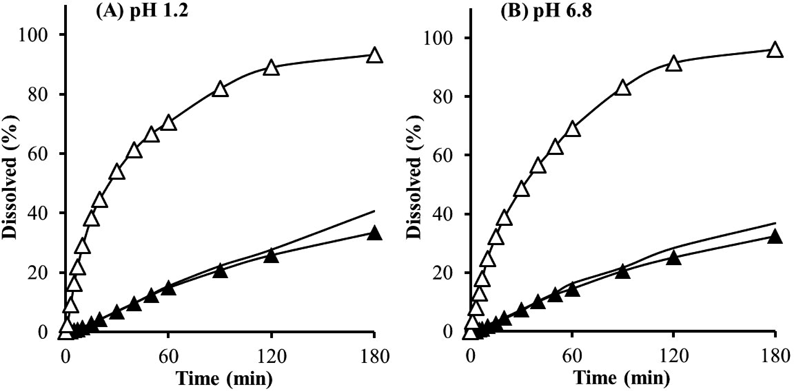
Dissolution experiments from cryo-milled particles and mechanical processed particles were conducted using the paddle method with a dissolution tester (NTR-3000, Toyama Sangyo Co., Ltd., Osaka, Japan), according to the Japanese Pharmacopeia, 17th edition (JP17). Sample powders corresponding to 10 mg Phe were placed into 900 mL of the dissolution media while maintaining the temperature at 37 ± 0.5 °C. The rotation speed of the paddle was set to 50 rpm. The first fluid (pH 1.2) and second fluid (pH 6.8) defined in the dissolution test of JP17 were used to estimate the dissolution behavior in the gastrointestinal fluid. The tests were performed under sink conditions in both media. Aliquots (1 mL) of the solution were taken at 1, 3, 5, 7, 10, 15, 20, 30, 40, 50, 60, 90, 120, and 180 min and filtered through a hydrophilic PTFE membrane filter with a 0.20-µm pore size (Dismic-13HP, Advantec Toyo). Subsequently, the same volume of fresh medium was added to the dissolution vessel to maintain the original volume. Drug concentration in the filtrates was determined by HPLC as described above. Dissolution profiles of the original Phe powder were also determined as a reference.
Micromeritic PropertiesThe micromeritic properties of the cryo-milled particles and the mechanical processed particles were evaluated using a multifunctional powder property measuring instrument (Multi-tester MT-1000, Seishin Enterprise Co., Ltd.), according to the general tests of JP17. The angle of repose was used to assess flowability (flowing performance of the powder). The sample powder was placed onto a circular stage with a fixed diameter (8 cmϕ) through a funnel while vibrating to form a symmetrical conical powder bed while varying the height of the funnel. The funnel position was kept approximately 3 cm above the top of the powder pile as it was being formed to minimize the impact of falling powder on the top of the cone. The diameter and height of the conical powder bed were measured using a cathetometer (Travelling microscope, Shimadzu Co., Ltd.). The angle of repose (α) was calculated from Eq. (4):
 | (4) |
The packability (packing performance of the powder) was evaluated using the density of the powder bed. The sample powder was gravimetrically filled into a measuring cylinder (22.5 mm diameter and 10-mL capacity) through the funnel in same manner described above. The bulk density (ρbulk), indicating the loosest density, was determined from the weight of filled powder. Then, the cylinder was tapped to pack the powder until no further volume change occurred (500 taps). The tap density (ρtapped), indicating the closest density, was determined from the final tap volume of the powder. The compressibility index was calculated using Eq. (5):
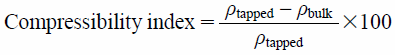 | (5) |
Results and Discussion
Ultra Cryo-MillingPhe particles were bead-milled in LN2 under cryogenic conditions using the ultra cryo-milling technique.6) The morphological appearance of the ultra cryo-milled particles of Phe and PEG is shown in Fig. 1, along with intact particles as a control. The intact Phe appeared as block-like or plate-like crystals with a smooth surface and size of about 10–20 µm (A-1). Intact PEG particles were plate-like fragments with a size of about 100 µm (A-2). The milled Phe particles (M-Phe, A-3) were pulverized to sub-micron size in the primary particle level to form an aggregated product, as previously reported.12) However, the co-milled particles of Phe and PEG (M-(Phe + PEG), A-4) were also pulverized to sub-micron size in the primary particle level. The PEG particles were not pulverized as finely as the Phe particles, and the fine particles of pulverized Phe appeared to form aggregates around larger PEG fragments. The co-existence of PEG in the cryo-milling process would be advantageous to improve the mixing uniformity between fine drug and PEG particles in primary level as shown in Fig. 1 (A-4). In addition, the pulverized fine PEG particles would effectively avoid the placebo massive aggregates of sole PEG in the next dry powder processing step (described in next section). Their PSD results demonstrated that the PSD curve of M-Phe shifted to the fine range and the particles became finer than those of intact Phe particles, as shown in Fig. 2. In contrast, the PSD curve of M-(Phe + PEG) overlapped with that of intact Phe and intact PEG, but the Phe particles appeared to be pulverized to fine particles as much as M-Phe based on the SEM images. Most of the PEG particles were pulverized to 40 µm or less and formed strong aggregates with the fine Phe particles due to their adhesive characteristics. It was suggested that the aggregated particles were not sufficiently dispersed in compressed air during the size measurement.
The dissolution profiles of the ultra cryo-milled particles and intact Phe particles are shown in Fig. 3. The M-Phe particles had a slower dissolution profile equivalent to intact Phe. Thus, the hydrophobic Phe particles were speculated to form strong secondary aggregates in the aqueous phase despite their primary submicron size, resulting in poor dispersion in the aqueous phase.13,14) In contrast, M-(Phe + PEG) showed a significant improvement in dissolution profile in both fluids compared to intact Phe and M-Phe. The pulverized Phe particles also formed secondary aggregates with PEG segments after co-milling, as shown in Fig. 1. However, hydrophilic PEG improved the wettability of whole powder and facilitated penetration of water between Phe particles. The remarkable enlargement of effective surface area related to micronization was another factor that contributed to their increased dissolution.
Ultra cryo-milling using LN2 is a hybrid milling method providing advantages of both wet and dry milling. The extent of miniaturization is equivalent to that of conventional wet milling in water. However, a drying process is not required after milling due to spontaneous evaporation of LN2 under ambient temperature and pressure. Therefore, although considered a wet method, the process can be treated as a dry method. Results also confirmed that the drug particles maintained their original crystalline form after milling (data not shown). The SEM observations and PSD results suggest that the co-milled particles from this method tended to form strong aggregates of fine particles. Co-milling with water-soluble PEG could effectively solve the reduced dissolution improvement attributed to secondary aggregation, but the co-milled products still had the problem of poor powder handling. Such inferior micromeritic properties would be troublesome in later manufacturing (e.g., mixing, granulation, tableting). Therefore, additional particle designs were investigated to improve the micromeritics of the cryo-milled particles.
Effect of Core Particles on Mechanical Powder ProcessingDry powder processing using a mechanical powder processor has been developed by applying CS and modified CS particles as a core.9,15) Considering that core particles larger than CS are more effective for improving powder micromeritics, NP (approx. 100 µm) and CP (approx. 250 µm) particles were used as the core particles in the present study. The loading volume of powder in the machine is a key factor for successfully dry coating around core particles based on the ordered mixing principle. For CS, approximately 30 mL of total bulk volume was optimum to provide effective shear stress. When dry powder processing was performed using NP or CP as the core, the mechanical powder processor operated under the same conditions as CS was overloaded, and the process could not be completed. Since the clearance between the blade and inner wall (1 mm) was considered to be too small for larger NP and CP particles, powder got clogged between them. Therefore, to complete the dry composite mixing process successfully for NP and CP, the loading volume was reduced from 30 mL to 20 mL. This decrease in loading volume was also important for maintaining the agglomerated forms of NP and CP spheres, which were easily broken under high-sheared stress.
The resulting particles of CS, NP, and CP with M-Phe prepared through mechanical powder processing are referred to as CS9 : 1 : 0, NP9 : 1 : 0, and CP9 : 1 : 0, respectively, and those with M-(Phe + PEG) are referred to as CS9 : 1 : 1, NP9 : 1 : 1, and CP9 : 1 : 1, respectively. The morphological appearance of the particles are shown in Figs. 4–6. The magnification ratio increased from left to right. Photographs showed that intact CS particles were grain-like, approximately 20 µm in diameter, and that its surface was smooth (Fig. 4, B-1). For CS9 : 1 : 0 and CS9 : 1 : 1 subjected to mechanical powder processing with milled products, the low-magnification images suggested that their particle size was equivalent to that of intact CS (Figs. 4, B-2, B-3), which was also supported by size distribution results. Three size distribution curves (intact CS, CS9 : 1 : 0, CS9 : 1 : 1) nearly overlapped and contained no peak derived from milled Phe (Fig. 7). However, the middle- and high-magnification photos showed that the surface of the processed particles had clearly changed to a mosaic pattern. The surface of PEG-free processed particles (CS9 : 1 : 0), especially, were coated with numerous fine fragments of submicron to single micron size (Fig. 4, B-2), which were identical to M-Phe particles in size. This morphological surface feature was also observed in the processed particles of both NP and CP cores (NP9 : 1 : 0 and CP9 : 1 : 0, shown in Figs. 5-C-2 and 6-D-2, respectively). These results indicated that the cryo-milled fine Phe particles (M-Phe) had adhered to the surface of all types of core particles, irrespective of core size. As a result, the ordered mixture-type composite particles having CS/NP/CP as a core particle and Phe as a coating layer were prepared successfully through co-processing milled Phe and three types of core particles. For NP and CP, the agglomerated granules were never crushed by high shearing stress during operation. Thus, these composite particles with an ordered mixture-type structure are called “OM particles.”
For co-processed particles with PEG (9 : 1 : 1 series, PEG-in), SEM observations and particle size distribution curves indicate that the sizes of processed particles were nearly identical to those of PEG-free OM particles in all three cases (CS, NP, CP). Such results demonstrated that the agglomeration or crushing of spherical cores did not occur during the process, even for waxy material. In only CS case, the processed particles (CS9 : 1 : 1) were detected as slightly smaller size than the core particles (CS). Such phenomenon has been also reported in our previous paper.9) The reason why the reversal phenomenon occur is not clear, but the surface property of PEG-coated OM particles might avoid the soft aggregation of intact CS particles with adhered moisture on their surface. In contrast, the surface structure of co-processed PEG-in particles (9 : 1 : 1) was clearly different from that of PEG-free particles (9 : 1 : 0) in all three cases. For PEG-in series (9 : 1 : 1), the fine fragments observed on the surface of PEG-free particles (9 : 1 : 0) were partially fused (CS) or almost nonexistent (NP, CP), with an uneven and smooth surface in all three cases (Figs. 4-B-3, 5-C-3, 6-D-3). This characteristic structure was attributed to waxy PEG with a low melting point (62–65 °C). The PEG particles were partially melted by the instantaneous heat generated locally by high-shear stress between the rotating blades and the inner round wall of the vessel. Thus, the core particles were coated with a partially melted PEG layer in which the milled Phe particles were embedded. The operation was run under a controlled temperature by circulating water at 20 °C in a jacket around the vessel to maintain a temperature below 30 °C. When the operation was conducted under circulating water at 50 °C in a jacket around the vessel, almost all of the powder agglomerated to form a large massive lump or to become stuck to the vessel wall and blades. Therefore, partial melting of PEG particles under an appropriate temperature is a key factor for successfully coating core particles with M-(Phe + PEG) and with minimum loss.
The PSD of NP-based and CP-based OM particles also exhibited a single peak corresponding to that of the core particles (Fig. 7), similar to the CS-based OM particles. These results demonstrate that the agglomerated granules (NP, CP) composed of mannitol or crystalline cellulose maintained their original size and shape and were not broken under high-shear stress. A previous report indicated that intact (unmilled) drug particles subjected to mechanical powder processing were pulverized effectively and layered onto CS core particles.9) The CS particles had elastic properties under mechanical stress and could also act as milling-assistant media to break the brittle drug particles as well as its main role as a core. Thus, milling and ordered mixing processes simultaneously progress within a short time to form OM particles.9) This method of OM preparation is categorized as a break-down type, which requires sufficient mechanical strength of the core particles so they can withstand the high mechanical stress needed to conduct pulverization and powder coating simultaneously. However, NP and CP, the agglomerated granules of mannitol and crystalline cellulose, respectively, did not have the strength sufficient to be milling-assistant media such as CS, especially in NP. Therefore, the operational conditions needed to be different than those previously described. The drug particles needed to be pre-milled by some method prior to co-processing with NP and CP core particles. In other words, NP-based and CP-based OM particles could be prepared because pre-milled drug particles were applied to dry powder processing in this study. Paradoxically, successful ordered mixing with NP or CP core particles requires cryo-milling of the drug particles in advance. Thus, the NP- and CP-based OM particles were obtained by depositing pre-micronized Phe particles on the surface of the core particles. The preparation procedure using the dry processor is categorized as a build-up type in this case. The build-up process combined with cryo-milling should be widely applicable because of the independence of the drug and core particles and the mechanical properties. In addition, NP and CP are frequently used in commercial products to prepare coated fine granules for controlled release, such as enteric release or taste masking.16–18) These results suggest that spherical drug granules of various sizes can be prepared using a dry processing technique with a mechanical powder processor. The appropriate size of core particle should be selected according to their application (e.g. for tableting, for bitter-taste masking particle).
Total recovery through the dry composite mixing process was 93 wt% or more against the loaded powders (Table 4). The drug contents in the recovered products were also analyzed by HPLC. The recovered products were placed on a sieve with 45-µm openings to separate free drug particles not rigidly adhered to the core particles. Both samples were analyzed after recovery and sieving, but CS-based OM products were too small to be separated using the sieve. In the PEG-free OMs series (CS9 : 1 : 0, NP9 : 1 : 0, CP9 : 1 : 0), the Phe recovery tended to decrease as the diameter of core particles increased (98.00, 81.50, 75.20%). The larger the core particles, the smaller the specific surface area per unit weight, resulting in a decrease in the total area available for adherence by guest particles.4) In contrast, in the PEG-in OMs series (CS9 : 1 : 1, NP9 : 1 : 1, CP9 : 1 : 1), the Phe recovery was similar for all three products (82.40, 83.17, 85.48%), indicating that the entrapment efficiency of Phe on the surface of all three types of core particles was approximately equivalent. These results are not consistent with those based on the surface area of PEG-free OMs, perhaps because the partially melted PEG acted as a dry binder to promote adhesion/fixation of M-Phe onto the core. The retention ratio, which indicates the remaining percentage of the drug on the surface of the core particles after sieving (i.e., the index of adhesion strength between Phe and core), increased for both the NP (78.16→84.92%) and CP series (85.90→93.69%) upon co-formulating with PEG. These results indicate that powder processing in the presence of PEG facilitates drug deposition on the core particles. However, for the CS series, melted PEG might induce agglomeration/coalescence among smaller CS particles, resulting in inferior formation of OM particles (98.00 vs. 82.40%). The supportive effect of PEG on formation of the composite particles with a mononuclear structure was more advantageous when the core size was large enough.
Table 4. Recovery and Phenytoin Content in Mechanical Processed Composite Particles
| Symbol | Total recoverya) | Phe content (mg/100 mg) | Phe recovery (%)e) | Retention ratio (%)f) | Number of coating layersg) |
|---|
| Theoreticalb) | Measuredc) | Measured on sieved) |
|---|
| CS9 : 1 : 0 | 93.12% | 10.00 | 9.80 | — | 98.00% | — | — |
| CS9 : 1 : 1 | 95.61% | 9.09 | 7.49 | — | 82.40% | — | — |
| NP9 : 1 : 0 | 95.54% | 10.00 | 8.15 | 6.37 | 81.50% | 78.16% | 1.27 |
| NP9 : 1 : 1 | 96.38% | 9.09 | 7.56 | 6.42 | 83.17% | 84.92% | 1.28 |
| NP6 : 1 : 1 | 96.06% | 12.50 | 10.49 | 8.48 | 83.92% | 80.84% | 1.70 |
| NP3 : 1 : 1 | 93.62% | 20.00 | 17.69 | 12.34 | 88.45% | 69.76% | 2.47 |
| CP9 : 1 : 0 | 95.36% | 10.00 | 7.52 | 6.46 | 75.20% | 85.90% | — |
| CP9 : 1 : 1 | 97.25% | 9.09 | 7.77 | 7.28 | 85.48% | 93.69% | — |
a) Total recovery = (recovery weight/loading weight) × 100. b) Theoretical: Drug content was calculated from the loading weight of each material. c) Measured: Drug content in the processed particles was determined using HPLC. d) Measured on sieve: Drug content in the processed particles remaining on the sieve (45-µm opening) was determined using HPLC. e) Phe recovery (%) = (measured Phe content/theoretical Phe content) × 100. f) Retention ratio (%) = (measured residual Phe content on sieve/measured Phe content) × 100, indicating the percentage of Phe remaining adhered to the core particles after sieving. g) Number of coating layers = measured residual Phe content on sieve/arithmetic Phe content for a monolayer coating (5.00 mg/100 mg of NP).
To investigate the effect of the loading ratio of drug to core particles, the OM particles were prepared using a greater loading amount of M-(Phe + PEG) only in the NP series, as shown in Table 3. The loading ratio of NP : M-(Phe + PEG) was varied from 9 : 2 to 6 : 2 to 3 : 2. As a result, 9.09% of theoretical Phe content in NP9 : 1 : 1 increased to 12.50 and 20.00% in NP6 : 1 : 1 and NP3 : 1 : 1, respectively. The appearance of resulting products is shown in Fig. 8. Unlike the photographs of NP9 : 1 : 1 shown in Fig. 5-C-3, the fine free particles apart from the OM body are shown in Figs. 8-C-4 and 8-C-5 despite co-loading with PEG. The free fraction apparently increased with Phe content. These free particles are unlikely to be fragments generated from broken core NP granules by shear stress, for reasons previously discussed. The particles were considered to be free or softly gathered M-(Phe + PEG) and could not adhere onto the surface of the core particles. The smooth surface of NP9 : 1 : 1 covered with fused PEG (Fig. 5-C-3) gradually became rough with tiny protrusions in NP6 : 1 : 1 (Fig. 8-C-4). This characteristic morphology was remarkable in NP3 : 1 : 1 (Fig. 8-C-5). These results indicate that a drug loading greater than 10% is too much for adhering tightly onto NP core particles. The drug contents of 3 levels of NP-based OM particles were determined to evaluate drug adherence quantitatively (Table 4). The measured Phe content increased with loading amount of M-(Phe + PEG) (7.56→10.49→17.69 mg/100 mg OM particles in NP9 : 1 : 1, NP6 : 1 : 1 and NP3 : 1 : 1, respectively) while maintaining a greater percentage of Phe recovery (83.17, 83.92, 88.45%). However, the retention ratio (i.e., the amount of Phe particles remaining on core particles under air-flow stress during sieving) decreased (84.92→80.84→69.76%). As previously mentioned, this decrease in retention ratio indicates that the drug particles are less likely to adhere to the core surface in higher drug loading. These results are consistent with the SEM observations (Fig. 8). The tiny protruding drug particles were apparently detached from the core particles after sieving.
To examine the coating structure in detail, the theoretical Phe content (mg/100 mg), assuming a monolayer coating, was calculated according to a calculation formula (6) and (7) published previously by Yang et al.19) These formula assume that both host (NP) and guest (M-Phe) particles are perfect spheres with a uniform particle size (mono-dispersion) and do not deform during the coating process, and that the surface of the host particle was completely covered with a single layer of guest particles in the closest packing manner (100% surface coverage). In this case, the diameter of intact NP particles was set to 97.85 µm based on the measured value of D50 (Fig. 7). On the other hand, the diameter of the M-Phe particles was considered as 1.00 µm based on SEM observations (Fig. 1). The average size (2.46 µm, Fig. 2) measured by laser diffractometry would not reflect the real single particle size because they must be aggregated during measurement. In addition, the true density of Phe and NP was set to 1.40 and 1.11 g/cm3, respectively, which were measured values through helium replacement method using a pycnometer in this study. Based on these assumptions, Phe content of monolayer (wt%) is:
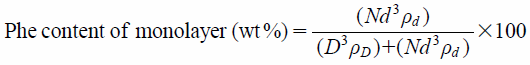 | (6) |
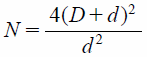 | (7) |
where
Host (NP) particle diameter (D): 97.85 µm
Guest (Phe) particle diameter (d): 1.00 µm
Host (NP) true density (ρD): 1.11 g/cm3
Guest (Phe) true density (ρd): 1.40 g/cm3
By substituting above data into the formula (6) and (7), the Phe content deposited on the surface of NP core was calculated to be 5.00 wt% (=5.00/100 mg). Thus, if the Phe content of the OM was found to be 5.00/100 mg or 10.00/100 mg, the composite particles are considered to be a monolayered or double-layered structure. From the Phe content measured in the residue on the sieve, the conceptual number of coating layers were calculated to be 1.28 layers in NP9 : 1 : 1, 1.70 layers in NP6 : 1 : 1, and 2.47 layers in NP3 : 1 : 1 (Table 4). Since the real conditions are different from the theoretical ones (e.g., the host/guest particles are not spherical and are not all exactly the same size) and the actual coating state is not accurately reflected, but these values could predict that the drug particles would form multi-particle layers. In proportion to the number of coating layer, it was also found that the particle size of OM particles became slightly larger (data not shown). That is, the size of the OM particles increased as thickening the coating layer of Phe particles around the core particles. The pre-milling of guest particles and the strong shearing force of the mechanical processor were thought to be the driving forces for creating a multi-layer coating.
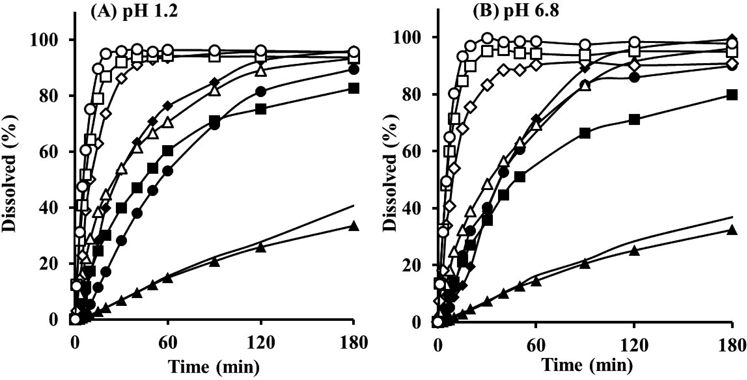
Pharmaceutical Performance of Composite ParticlesThe dissolution behavior of three series of OM and cryo-milled particles was investigated to estimate the solubilization effect. The OMs that dissolved rapidly in both fluids compared to intact Phe and M-Phe are shown in Fig. 9 (closed and open symbols indicate PEG-free and PEG-in series, respectively). In general, the milled particles of a poorly water-soluble drug with high hydrophobicity tended to flocculate in aqueous medium, resulting in poor improvement of dissolution profiles.13,14) The strong shear stress by the mechanical powder processor allowed the fine Phe particles to spread a thin layer over the surface of core particles, preventing inter-particle aggregation. The increased effective surface area related to wetting improved dissolution of PEG-free OM particles (CS9 : 1 : 0, NP9 : 1 : 0, CP9 : 1 : 0, closed symbols in Fig. 9) because the thin drug layer was composed of fine drug particles. For PEG-in OM particles (CS9 : 1 : 1, NP9 : 1 : 1, CP9 : 1 : 1, open symbols in Fig. 9), the dissolution of each type of OM particle was enhanced further with rapid onset compared to that of PEG-free OM particles. Hydrophilic PEG incorporated in the outer Phe layer likely improved the wettability of OM particles and promoted spontaneous detachment of the drug particles from the surface of the core particles in aqueous phase. The difference of release behaviors between PEG-free (9 : 1 : 0) and PEG-in (9 : 1 : 1) OM particles would not be attributed to primary particle size of Phe as previously shown in appearance (Fig. 1). These results demonstrated that the improved dissolution behavior of OM particles was due to both wide spreading and rapid wetting of Phe particles (both morphological and functional effects). Focusing on the type of core particle, dissolution of NP-based OM occurred most rapidly among OMs. The NP particles composed of mannitol were the only water-soluble core particles. The dissolution of core particles accelerated the penetration of water into the drug layer and dispersion/dissolution of drug particles.
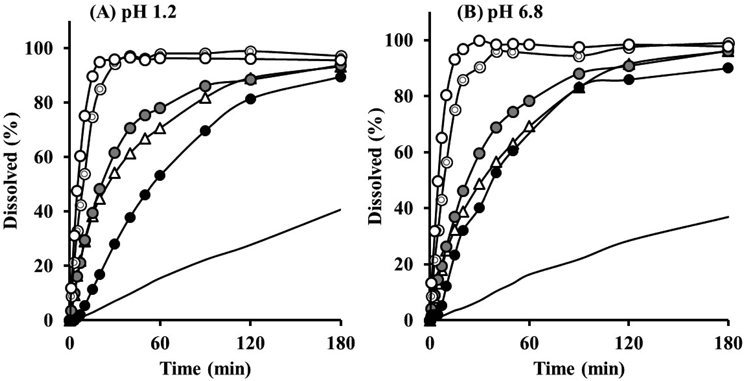
The dissolution behavior of NP-based OM particles (NP9 : 1 : 1, NP6 : 1 : 1, NP3 : 1 : 1) was also investigated through the composition ratio of core/drug. The rapid dissolution behavior of NP9 : 1 : 1 was decreased as Phe content in OM particles increased, despite co-formulating with water-soluble PEG (Fig. 10). The poor dissolution performance at greater drug concentrations, especially in NP3 : 1 : 1, was attributed to the thickened drug layer around the core as shown in Table 4 (number of coating layers). The multiple layering of M-(Phe + PEG) was assumed to disturb the penetration of water into the core, delaying the fine dispersion of milled Phe particles. However, the delay was not serious compared to that of the CS-based OM particles reported previously.9) NP is composed of water-soluble mannitol, which would be main reason why the dissolution of Phe was not so delayed. The high PEG content would be another factor maintaining moderate solubilization effect in NP3 : 1 : 1. The results suggested that the blending ratio was a critical factor for designing OM particles with enhanced dissolution properties. In addition, no change in the X-ray powder diffractogram after mechanical powder processing clearly revealed that the crystalline form and crystallinity of Phe was not different from the original (data not shown). Thus, improvement in dissolution was not derived from crystalline properties such as amorphism or polymorphism, suggesting outstanding storage stability. The sequential process of cryo-milling and dry powder processing technique could be applied to thermally unstable drugs. In case that the dry powder processing operation is performed under heating (50 °C or higher), the chemical and physicochemical (e.g. crystalline) change of drugs should be evaluated. This improvement in dissolution behavior was not derived from molecular or crystalline features, but from morphological features such as a spread of cryo-milled fine particles on the surface of the core particles. The sequential process of cryo-milling and dry composite mixing is an alternative approach for improving the dissolution characteristics of poorly water-soluble drugs in a manner different from chemical (complexation including inclusion compounds) and crystalline approaches (solid dispersion, self-emulsification).
Micromeritic Properties of Composite ParticlesFlowability and packability are the most important characteristics for determining powder processability in the manufacturing of oral solid dosage forms. In general, pharmaceutical powders with poor handling properties can be improved by mixing with a lubricant, such as silica or magnesium stearate, which coat the base particle.20) Jallo et al. reported that dry coating using a magnetically assisted impaction coating (MAIC) technique effectively broke down an agglomerated mass of ibuprofen particles by coating the surface of the individual particles with nano-silica.21) Abdullah and Geldart reported that silica-coated powders could induce smaller adhesion forces between particles that collapsed as the particles settled on each other, improving flowability and packability.22) The improvement in flow of the dry coated powders was due to the presence of the spherical silica on the surface of particles.19,23) The presence of these nanoparticles on the surface of the host particles changes the geometrical position between the larger particles that reduces the contact area, decreasing the adhesion forces and improving flow properties.23–26)
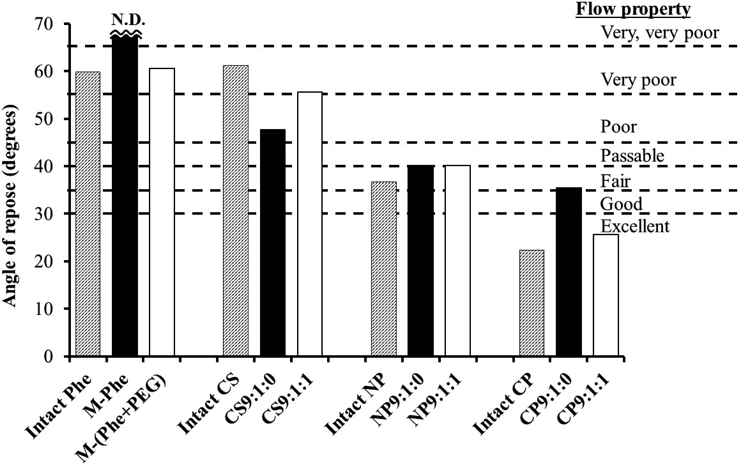
The powder flow properties of OM particles manufactured by mechanical powder processing were evaluated using the angle of repose (AOR) of a conical pile of powder. The measured values are shown in Fig. 11 along with the flow properties proposed by Carr.27) The gray, black, and white bars indicate intact powder, PEG-free particles, and PEG-in particles, respectively. The AOR values are determined by the strength of the adhesion force and the type of friction at the points of contact between the particles. Thus, for powders with poor flowability, the larger adhesion force between the particles results in sticking, producing steeper slopes and higher AOR values. Intact Phe and milled powder [M-Phe, M-(Phe + PEG)] had greater AOR values, indicating that they had poor flow properties (i.e., were highly cohesive powders). The M-Phe, especially, was difficult to feed through the funnel because of its strong adhesion property. Results showed that the AOR value was reduced for all OMs; the extent of reduction was dependent on the size of the core particles. The NP- and CP-based OM particles exhibited improved flowability (“passable” to “fair” for NP-based OM, “fair” to “excellent” for CP-based OM), compared to cryo-milled powder [M-Phe and M-(Phe + PEG)]. However, CS-based OM particles remained in the “poor” region. Jallo et al. reported that flowability could not be improved unless the particle size was greater than 30 µm.21) Therefore, the flowability of CS-based OM particles with a value of approx. 20 µm in D50 could not be improved, even after processing. These results indicate that improvement in flowability is due to size enlargement, not to surface modifications of processed particles.
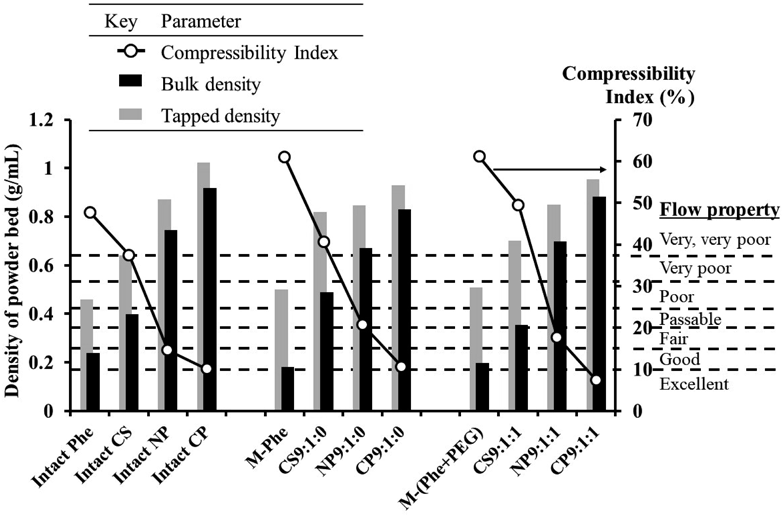
Along with flowability, packability is an important factor in evaluating the filling performance of powder (e.g., filling of the die for tableting or capsule filling operation). The loosest and closest densities of sample powder beds were measured before and after tapping, and the compressibility index (CI) was calculated to assess packability. Densities are shown in Fig. 12, in which the CI values are co-plotted. Carr’s guide of flow properties27) are also indicated as horizontal lines. Three groups (intact, PEG-free, PEG-in) are arranged in order from left to right. Thus, powder samples with a larger particle size tended to provide a powder bed with greater density. As a result, highly adhesive, milled powders [M-Phe, M-(Phe + PEG)] with a CI value greater than 60% had the value reduced after processing. The flow properties of NP- and CP-based OM particles were categorized as “passable” to “excellent” for OM samples. The CS-based OM particles were ranked “very, very poor.” For very small particles, van der Waals forces between the particles would exceed the gravity of the particle, inducing interparticle aggregation. When comparing the PEG-free and PEG-in series, no significant difference was found in their CI values. The inferior micromeritic properties of the pulverized particles were improved by formation of composite particles with a large core. Further improvement in flowability is expected by co-formulating fine particles, such as silica, during processing. Results demonstrated that the particles processed with an ordered mixture-structure had the additional advantage of good micromeritics properties (flowability and packability) while maintaining improved release behavior.
Conclusion
Poorly water-soluble Phe particles were milled or co-milled with PEG in LN2. The milled products were well micronized on the single-micron to submicron level. In addition, designs of cryo-milled drug particles were investigated to improve micromeritics properties. The cryo-milled particles were mechanically processed with a core particle having a round/grain shape and a larger size. CS particles, NP granules, and microcrystalline CP granules of different sizes (small, medium, and large, respectively) were used. The morphological appearance and size distribution indicated that the co-processed particles were spherical composite particles with a core-shell structure. The composite particles were called “OM” particles after the ordered mixture style. The cryo-milled fine drug particles were deposited on the surface of core particles, both PEG-free and PEG-in. Co-milled PEG was also fixed on the surface of core particles, with the drug particles embedded in a partially melted PEG layer. Through sequential steps of cryo-milling and mechanical powder processing, the composite OM particles with a double structure could be realized, in which the composite particles (secondary particles) consisted of core particles covered with the finely milled particles (primary particles).
The dissolution behavior of OM particles was strongly dependent on the primary structure. Fine drug particles widely spread over the surface of the core particles immediately dispersed in aqueous phase, promoting the rapid release of the drug. In addition, co-micronized PEG enhanced the wettability of a hydrophobic pulverized drug layer, preventing flocculate formation and promoting rapid dissolution in the aqueous phase. In contrast, the micromeritics properties of OM particles were strongly related to the secondary structure. The flowability and packability of OM particles were dramatically improved based on the size of core particles, in contrast to the cryo-milled product.
In conclusion, designing the primary and secondary structures of OM could simultaneously improve the dissolution and powder properties of poorly soluble drugs. These results will be useful to improve the dissolution of poorly water-soluble drugs. In addition, the products are well-suited to preparing coated particles, which mask a bitter taste or improve swallowability, by embedding in orally disintegrating (OD) tablets due to their spherical and uniform size distribution. The cost effectiveness should be discussed in the future from the view point of LN2 price for practical use of pharmaceutical industry. Further, the contamination issue generated from zirconia beads in cryo-milling process should be argued toward practical production. The cryo-milling in LN2 using dry ice beads in replace of zirconia beads (novel contamination-free milling) would be one of the promising countermeasures as reported by authors.28,29)
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Fagerberg J. H., Bergstroem C. A. S., Ther. Deliv., 6, 935–959 (2015).
- 2) Amidon G. L., Lennernäs H., Shah V. P., Crison J. R. A., Pharm. Res., 12, 413–420 (1995).
- 3) Leleux J., Williams R. O. 3rd, Drug Dev. Ind. Pharm., 40, 289–300 (2014).
- 4) Noyes A. A., Whitney W. R., J. Am. Chem. Soc., 19, 930–934 (1897).
- 5) Jimbo G., “Recent Formulation Technologies and Their Application 3,” Iyaku Journal Inc., Tokyo, 1986, pp. 168–172.
- 6) Niwa T., Nakanishi Y., Danjo K., Eur. J. Pharm. Sci., 41, 78–85 (2010).
- 7) Brovik E. S., Sov. Phys., 5, 506–507 (1960).
- 8) Vasserman A.A., “Thermophysical Properties of Air and Air Components,” Izdatel’avto Nauska, Moscou, 1966, pp. 253–254.
- 9) Niwa T., Yoshida M., Hayashi N., Kondo K., Int. J. Pharm., 528, 624–636 (2017).
- 10) Buckley S. T., Frank K. J., Fricker G., Brandl M., Eur. J. Pharm. Sci., 50, 8–16 (2013).
- 11) Kondo K., Ito N., Niwa T., Danjo K., Int. J. Pharm., 453, 523–532 (2013).
- 12) Sugimoto S., Niwa T., Nakanishi Y., Danjo K., Chem. Pharm. Bull., 60, 325–333 (2012).
- 13) Yuminoki K., Seko F., Horii S., Takeuchi H., Teramoto K., Nakada Y., Hashimoto N., J. Pharm. Sci., 103, 3772–3781 (2014).
- 14) Zimper U., Aaltonen J., Krauel-Goellner K., Gordon K. C., Strachan C. J., Rades T., Pharmaceutics, 2, 419–431 (2010).
- 15) Saeki I., Kondo K., Nagase R., Yoshida M., Niwa T., JDDST, 54, 101281 (2019).
- 16) Shimizu T., Sugaya M., Nakano Y., Izutsu D., Mizukami Y., Okochi K., Tabata T., Hamaguchi N., Igari Y., Chem. Pharm. Bull., 51, 1121–1127 (2003).
- 17) Tasaki H., Yoshida T., Maeda A., Katsuma M., Sako K., Int. J. Pharm., 376, 13–21 (2009).
- 18) Maeda A., Shinoda T., Ito N., Baba K., Oku N., Mizumoto T., Int. J. Pharm., 408, 84–90 (2011).
- 19) Yang J., Sliva A., Banerjee A., Dave R., Pfeffer R., Powder Technol., 158, 21–33 (2005).
- 20) Zhou Q., Qu L., Gengenbach T., Denman J. A., Larson I., Stewart P. J., Morton D. A. V., Int. J. Pharm., 413, 36–43 (2011).
- 21) Jallo L. J., Ghoroi C., Gurumurthy L., Patel U., Dave R. N., Int. J. Pharm., 423, 213–225 (2012).
- 22) Abdullah E. C., Geldart D., Powder Technol., 102, 151–165 (1999).
- 23) Chen Y., Yang J., Dave R. N., Pfeffer R., AIChE J., 54, 104–121 (2008).
- 24) Chen Y., Jallo L., Quintanilla M. A. S., Dave R., Aspects, 361, 66–80 (2010).
- 25) Jallo L. J., Schoenitz M., Dreizin E. L., Dave R. N., Johnson C. E., Powder Technol., 204, 63–70 (2010).
- 26) Jallo L. J., Chen Y., Bowen J., Etzler F., Dave R. N., J. Adhes. Sci. Technol., 18, 367–384 (2011).
- 27) Carr R., Chem. Eng., 72, 163–168 (1965).
- 28) Sugimoto S., Niwa T., Nakanishi Y., Danjo K., Int. J. Pharm., 426, 162–169 (2012).
- 29) Uemoto Y., Toda S., Adachi A., Kondo K., Niwa T., Chem. Pharm. Bull., 66, 794–804 (2018).