Notes
Three New C19-Diterpenoid Alkaloids from Aconitum novoluridum
2021 年 69 巻 8 号 p. 811-816
詳細
2021 年 69 巻 8 号 p. 811-816
Three new aconitine-type C19-diterpenoid alkaloid namely novolunines A (1), B (2), and C (3), along with fifteen known diterpenoid alkaloids were isolated from the roots of Aconitum novoluridum, whose phytochemical investigations have never been reported before. The structures of three new alkaloids were established on the basis of spectra data (high-resolution electrospray ionization (HR-ESI)-MS, IR, one dimensional (1D)- and 2D-NMR). Noteworthily, novolunines A (1) and B (2) are two diterpenoid alkaloids bearing conformational isomerism. In addition, the diterpenoid alkaloids 1–3 did not show any anti-acetylcholinesterase (AChE) or anti-inflammatory activities.
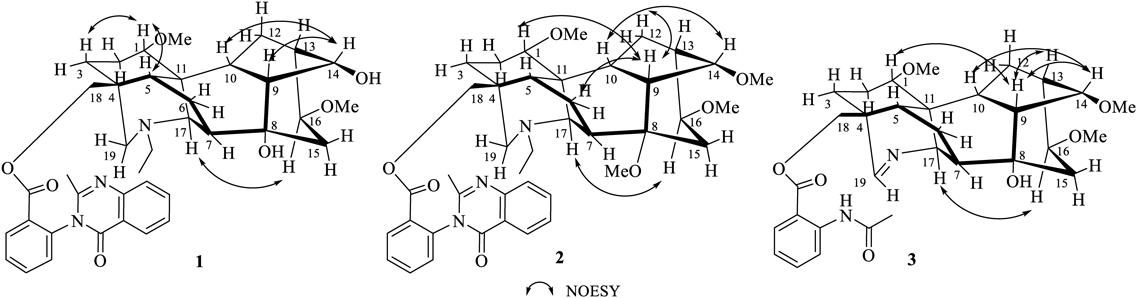
The Aconitum genus plants are known to contain a number of norditerpenoid and diterpenoid alkaloids.1) This kind of compounds have attracted considerable interest because of their complex structure, and noteworthy physiological effects.2) As our ongoing interest in the phytochemical studies on the pharmacologically interesting plants Aconitum and Delphinium, we obtained a series of structurally and chemotaxonomically interesting diterpenoid alkaloids.3–7) The plant Aconitum novoluridum Munz belongs to the Aconitum genus in the family of Ranunculaceae and is distributed in the northeast of Tibet in China, Bhutan, Nepal, and Sikkim at an attitude of 3800–4500 m.8) Aconitum novoluridum is similar to Aconitum apetalum, but the stem is lower and thinner, the leaves are smaller, the bracteoles are ovate, the sepals are purplish-red, and the petals are distinguished by short spines. The plant also plays an important role in the chemotaxonomy of Aconitum, which could be learned from the deep phytochemistry study on it.9) No phytochemical investigations appear to have been carried out on the constituents of Aconitum novoluridum, implying that the study is very important value for the chemotaxonomy of genera Aconitum. As part of an ongoing research program to investigate the chemistry diterpenoid alkaloids from Aconitum and Delphinium plants, we have carried out a study on the roots of Aconitum novoluridum, which led to the isolation three new aconitine-type C19-diterpenoid alkaloids, novolunines A–C (1–3), as well as fifteen known compounds, including eight aconitine-type, three lappaconitine-type C18-diterpenoid alkaloids, two ranaconine-type C18-diterpenoid alkaloid, and two denudatine-type C20-diterpenoid alkaloids have been isolated. Herein, we present the isolation and structural elucidation of alkaloids 1–3.
Column chromatography separation of the dichloromethane extract of Aconitum novoluridum afforded three new compounds novolunines A–C (1–3) as well as fifteen known diterpenoid alkaloids. The structures are shown in Fig. 1.

Novolunine A (1) was obtained as white amorphous powder. Its molecular formular was deduced to be C39H47N3O7 from a pseudomolecular ion at m/z 670.3483 in its high-resolution electrospray ionization (HR-ESI)-MS. The IR spectrum indicated the absorption peaks for a carbonyl group (1710 cm−1), and aromatic ring (1609, 1592, 1515, 1452 cm−1). The NMR spectrum of 1 exhibited characteristic features of the aconitine-type C19-diterpenoid alkaloids,10–15) bearing an N-ethyl group (δH 1.01, 3H, t, J = 7.2 Hz; δC 13.6 q, 49.3 t), and two methoxy groups (δH 3.19, 3.32, each 3H, s; δC 56.2 q, 56.4 q). Furthermore, the signals at δH 7.72 (1H, m), 7.26 (1H, m), 7.62 (1H, t, J = 8.0 Hz), 8.19 (1H, dd, J = 8.0, 1.2 Hz), 8.26 (1H, dd, J = 8.0, 1.2 Hz), 7.47 (1H, m), 7.77 (1H, m), 7.71 (1H, m), δC 153.9 s, 147.5 s, 137.6 s, 134.6 d, 134.0 d, 132.5 d, 129.8 d, 129.7 d, 128.6 s, 127.0 d, 127.0 d, 126.6 d, 120.8 s, a methyl group (δH 2.21 s; δC 24.1 q) and two carbonyl groups (δC 164.8 s, 162.1 s) indicated the presence of a 2-(2-methyl-4-oxoquinazolin-3-yl)benzoate moiety.16) The correlations in the heteronuclear multiple bond connectivity (HMBC) spectrum between H-18 [δH 3.91, 3.85 (each 1H, ABq, J = 11.2 Hz)] and carbonyl carbon at δC 164.8 revealed that the 2-(2-methyl-4-oxoquinazolin-3-yl)benzoate moiety is installed at the C-18. Comparison of the MS and NMR spectra of 1 with those of the known brevicanine A17) showed that these two compounds exhibited nearly identical 1H- and 13C-NMR resonances. The most obvious difference between compounds 1 and brevicanine A in their NMR spectra was the absence of a methoxy group in 1, thus validating the loss of 14 mass units in brevicanine A as found by mass spectrometry. The proton signal of H-14 was shifted downfield from δH 3.62 (1H, t, J = 4.8 Hz) in brevicanine A to δH 4.13 (1H, t, J = 5.2 Hz) in 1, along with its corresponding carbon signals was shifted upfield from δC 84.4 to δC 75.4, suggesting that the methoxy group at H-14 in brevicanine A was replaced by hydroxy group in 1. Except for this, the 1H- and 13C-NMR spectra of 1 and brevicanine A are very similar. Thus, the structure of 1 was determined as shown in Fig. 1, which were further confirmed by the analysis of its two-dimensional (2D) NMR data (Figs. 2, 3, Supplementary Materials).

The 1H-NMR and 13C-NMR signals of compound 1 in CDCl3 appeared doubling phenomenon at room temperature, indicating the natural isolated compound 1 existed conformers in CDCl3. Measurements of the 1H-NMR spectra in dimethyl sulfoxide (DMSO)-d6 at room temperature (25 °C) and high temperature (140 °C) were carried out to confirm the conformational isomerism of compound 1. As shown in Fig. 4, two sets of proton signals were appeared in DMSO-d6 at 25 °C (e.g., H-14 and H-14′, H-18A and H-18A′, respectively). Upon heating temperature to 140 °C, the doubling signals of hydrogen H-14, and H-18A were coalesced.
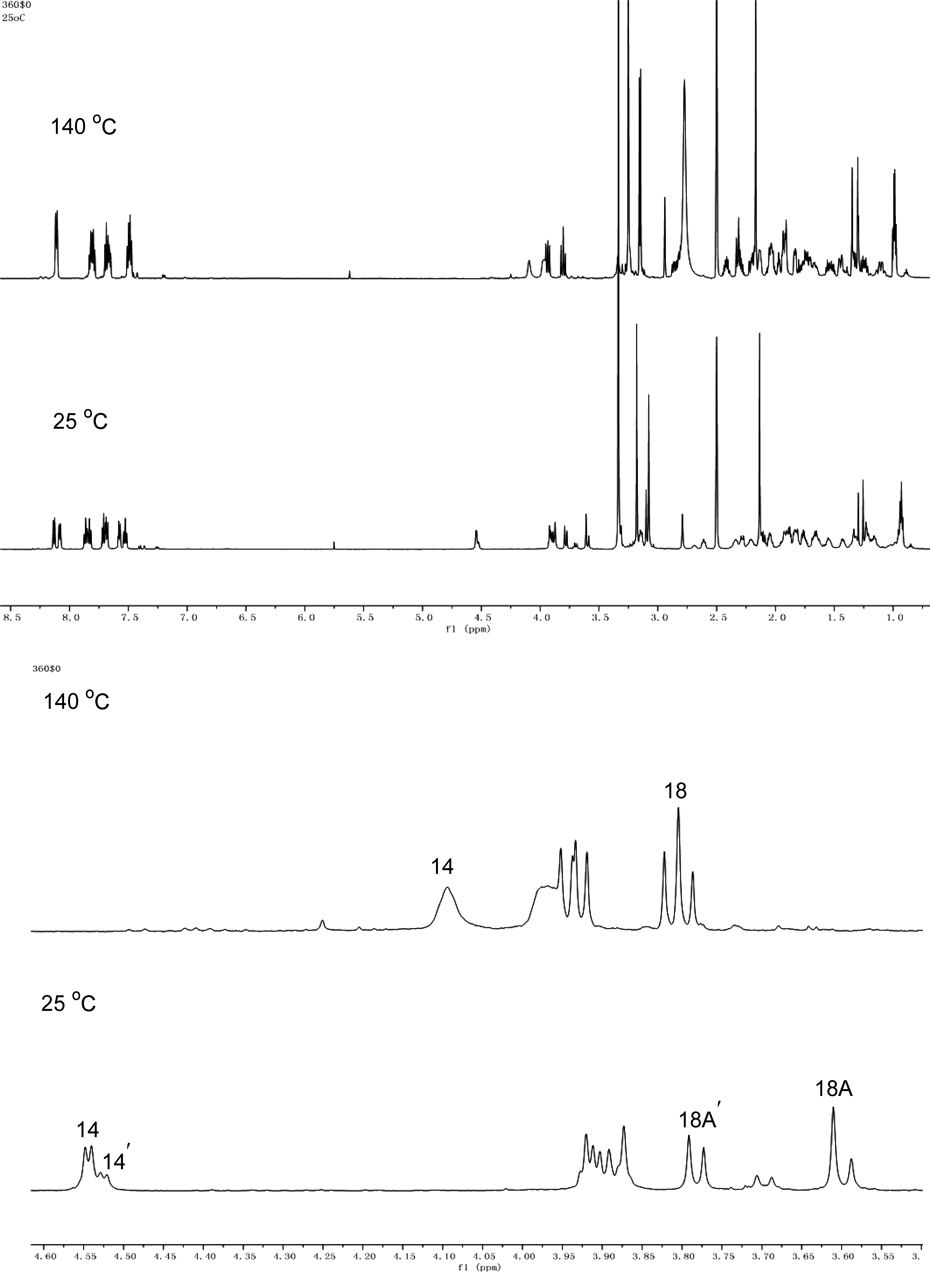
Novolunine B (2) was obtained as white amorphous powder. Its molecular formula was deduced as C41H51N3O7 on the basis of a protonated molecular ion at m/z 698.3794 [M + H]+ in the HRESIMS (calcd 698.3800). The IR spectrum displayed absorption bands characterized as ester carbonyl (1722 cm−1) and aromatic ring (1608, 1571, 1491, 1450 cm−1). The 1H-NMR spectrum showed the presence of an N-ethyl group (δH 0.97, 3H, t, J = 7.2 Hz; δH 2.39/2.26, 2H, m), four methoxy groups (δH 3.32, 3.28, 3.15, 3.01, each 3H, s), and a 2-(2-methyl-4-oxoquinazolin-3-yl)benzoate moiety [δH 8.15 (1H, d, J = 7.6 Hz), 7.57 (1H, t, J = 7.6 Hz), 7.68 (1H, t, J = 7.2 Hz), 7.22 (1H, m), 8.19 (1H, m), 7.41 (1H, t, J = 7.6 Hz), 7.72 (1H, m), 7.70 (1H, m), 2.21 (3H, s)]. The 13C and distortionless enhancement by polarization transfer (DEPT) NMR data demonstrated the existence of five methylene carbons (δC 28.7, 34.9, 70.7, 51.9, 48.4) and three quaternary carbons (δC 36.9, 76.9, 48.4). The aforementioned NMR features suggested that compound 2 is an aconitine-type C19-diterpenoid alkaloid with a substitution at C-18 position.10–15) The 2-(2-methyl-4-oxoquinazolin-3-yl)benzoate moiety was attributed to C-18 based on the long-range correlations between H-18 [δH 3.85/3.78 (each 1H, ABq, J = 11.2 Hz)] and a carbonyl carbon of 2-(2-methyl-4-oxoquinazolin-3-yl)benzoate moiety (δC 164.3) in its HMBC spectrum. The four methoxy groups could be readily assigned to C-1, C-8, C-14 and C-16 based on the cross-peaks between OCH3-1 and C-1, OCH3-8 and C-8, OCH3-14 and C-14, and OCH3-16 and C-16 observed in its HMBC spectrum (Fig. 2). The 13C-NMR data of 2 were very similar to those of known brevicanine D17) except for the methoxy group at C-8 in 2 was instead of ethoxy group in brevicanine D. Therefore, the structure of novolunine B was determined as 2. Moreover, the structure of 2 was confirmed by the analysis of its 1D and 2D-NMR data (Figs. 2, 3, Supplementary Materials). Similar to compound 1, compound 2 is also a diterpenoid alkaloid with conformational isomerism, which was confirmed by 1H-NMR spectrum in DMSO-d6 at 140 °C (Fig. S21–S23 in Supplementary Materials).
Novolunine C (3) was isolated as white amorphous powder. Its molecular formula was deduced to be C31H40N2O7 by HR-ESI-MS at m/z 553.2905 [M + H]+. The characteristic NMR data of 3 strongly suggested it to be an aconitine-type C19-diterpenoid alkaloid with a substitution at C-18 position.10–15) The NMR spectrum (Table 1) of 3 gave the distinctive signals at δH 3.39, 3.32, and 3.22 (each 3H, s), corresponding δC 57.7, 56.3, and 56.2 q for three methoxy group and δH 7.35 (1H, br s), δC 162.2 d for an imine group. The signal at δC 73.5 s indicated that it had one tertiary hydroxyl group. The absence of N-Et and the presence of a N = C (19)-H moiety in 3 suggested that it was an imine-containing alkaloid, like apetaldine G.7) This was further supported by the correlations of deshielded H-19 signal at δH 7.35 with C-5 (δC 41.5 d), C-17 (δC 62.2 d) and C-18 (δC 67.7 t) in the HMBC spectrum. Three methoxy groups were placed at C-1, C-14 and C-16 on the basis of HMBC correlations from OCH3-1 (δH 3.22, s) to C-1 (δC 82.5, d), OCH3-14 (δH 3.32, s) to C-14 (δC 85.2, d), and OCH3-16 (δH 3.329, s) to C-16 (δC 82.0, d), respectively (Fig. 2). In addition, the signals from an NH group [δH 10.98 (1H, br s)], an ortho-disubstituted benzene unit [δH 8.69 (1H, d, J = 8.4 Hz), 7.55 (1H, t, J = 8.0 Hz), 7.08 (1H, t, J = 8.0 Hz), 7.96 (1H, d, J = 7.8 Hz); δC 114.4 s, 141.7 s, 120.4 d, 134.9 d, 122.4 d, 130.4 d, 167.9 s], and an acetyl group (δH 2.23 s; δC 169.1 s, 25.5 q) indicated that compound 3 possessed an O-acetamidobenzoate moiety (−OCOC6H4-o-NHAc).18) The O-acetamidobenzoate moiety was located at C-18 on the basis of HMBC from H-18 (δH 4.38/4.34, each 1H, ABq, J = 11.4 Hz) to OCO-18 (δC 167.9, s). Thus, the structure of novolunine C (3) was assigned as 3 (Fig. 1), which was confirmed by extensive analysis of its 1D and 2D spectra (Figs. 2, 3, Supplementary Materials).
| No. | Compound 1 | Compound 2 | Compound 3 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 85.6 d | 2.81, dd (10.8, 6.4) | 84.4 d | 2.81, dd (9.2, 7.2) | 82.5 d | 3.28, m |
| 2 | 25.4 t | 2.03, m | 25.7 t | 2.07, m | 23.7 t | 1.77, m |
| 1.69, m | 1.69, m | 1.62, m | ||||
| 3 | 32.3 t | 1.54, m | 31.7 t | 1.46, m | 26.9 t | 1.75, m |
| 1.11, m | 1.13, m | 1.54, m | ||||
| 4 | 37.8 s | 36.9 s | 48.3 s | |||
| 5 | 45.3 d | 1.55, m | 44.7 d | 1.60, m | 41.5 d | 1.76 m |
| 6 | 24.6 t | 1.79, m | 23.5 t | 1.70, m | 25.2 t | 2.01, m |
| 1.27, m | 1.17, m | 1.51, m | ||||
| 7 | 45.5 d | 1.37, m | 39.3 d | 2.27, m | 52.8 d | 2.14, d (7.6) |
| 8 | 72.7 s | 76.9 s | 73.5 s | |||
| 9 | 46.7 d | 2.15, t (5.6) | 42.7 d | 2.09, m | 44.9 d | 2.18, t (4.8) |
| 10 | 37.5 d | 2.35, m | 37.9 d | 2.21, m | 44.8 d | 1.88, m |
| 11 | 48.5 s | 48.4 s | 49.1 s | |||
| 12 | 27.5 t | 1.79, m | 28.7 t | 2.25, m | 29.4 t | 1.86, m |
| 1.76, m | 1.72, m | 1.82, m | ||||
| 13 | 45.8 d | 2.02, m | 44.8 d | 1.60, m | 37.1 d | 2.40, t (5.6) |
| 14 | 75.4 d | 4.13, t (5.2) | 83.2 d | 3.46, m | 84.3 d | 3.72, t (4.8) |
| 15 | 38.2 t | 2.08, m | 34.9 t | 2.05, m | 41.1 t | 2.52, dd (15.2, 8.8) |
| 1.99, m | 1.93, m | 1.97, m | ||||
| 16 | 82.1 d | 3.38, d (7.6) | 83.1 d | 3.11, d (8.0) | 82.0 d | 3.25, m |
| 17 | 62.5 d | 3.08, s | 60.5 d | 2.68, s | 62.2 d | 3.88, s |
| 18 | 70.9 t | 3.91, ABq (11.2) | 70.7 t | 3.85, ABq (11.2) | 67.7 t | 4.38, ABq (11.2) |
| 3.85, ABq (11.2) | 3.78, ABq (11.2) | 4.34, ABq (11.2) | ||||
| 19 | 52.4 t | 2.42, m | 51.9 t | 2.33, m | 162.2 d | 7.35 br |
| 1.95, m | 1.86, m | |||||
| 21 | 49.3 t | 2.44, m | 48.4 t | 2.39, m | ||
| 2.32, m | 2.66, m | |||||
| 22 | 13.6 q | 1.01, t (7.2) | 13.0 q | 0.97, t (7.2) | ||
| OCH3-1 | 56.2 q | 3.19, s | 55.8 q | 3.15, s | 56.2 q | 3.22 s |
| OCH3-8 | 47.6 q | 3.01, s | ||||
| OCH3-14 | 57.1 q | 3.32, s | 56.3 q | 3.32 s | ||
| OCH3-16 | 56.4 q | 3.32, s | 55.6 q | 3.27, s | 57.7 q | 3.39 s |
| 1′ | 137.6 s | 137.0 s | 141.7 s | |||
| 2′ | 128.6 s | 128.0 s | 114.4 s | |||
| 3′ | 132.5 d | 8.19, dd (8.0, 1.2) | 131.9 d | 8.15, d (7.6) | 130.4 d | 7.96, d (8.0) |
| 4′ | 129.7 d | 7.26, m | 129.4 d | 7.57, t (7.6) | 122.4 d | 7.08, t (8.0) |
| 5′ | 129.8 d | 7.62, t (8.0) | 126.4 d | 7.68, t (7.2) | 134.9 d | 7.55, t (8.0) |
| 6′ | 134.6 d | 7.72, m | 133.9 d | 7.22, m | 130.4 d | 7.96, d (8.0) |
| 7′ | 164.8 s | 164.3 s | 167.9 s | |||
| 8′ | 169.1 s | |||||
| 9′ | 25.5 q | 2.23 s | ||||
| NH | 10.98 s | |||||
| 2″ | 153.9 s | 153.4 s | ||||
| 4″ | 162.1 s | 161.4 s | ||||
| 5″ | 127.0 d | 8.26, dd (8.0, 1.2) | 126.4 d | 8.19, m | ||
| 6″ | 126.6 d | 7.47, m | 126.0 d | 7.41, t (7.6) | ||
| 7″ | 134.0 d | 7.77, m | 133.9 d | 7.72, m | ||
| 8″ | 127.0 d | 7.71, m | 126.4 d | 7.70, m | ||
| 9″ | 147.5 s | 147.1 s | ||||
| 10″ | 120.8 s | 120.3 s | ||||
| CH3-2″ | 24.1 q | 2.21, s | 23.6 q | 2.20 s | ||
Structures of the fifteen known diterpenoid alkaloids were identified by spectral data and compared the values with reported literatures as cammaconine (4),19) acobretine C (5),20) acobretine D (6),21) aconorine (7),22) scaconitine (8),23) brochyponine A (9),24) brevicanine (10),25) N-deacetylscaconitine (11),23) excelsine (12),26) 8-O-acetylexcelsine (13),27) kirisine A (14),6) leucostonine (15),28) neofinaconitine (16),29) lepetine (17),30) and lepenine (18)31) (Fig. 1).
Compounds 1–3 were screened for their acetylcholinesterase inhibitory activity, using huperzine A as positive drug. However, none of them showed anti-acetylcholinesterase (AChE) activity (Table S1). Moreover, compounds 1–3 were also tested for their anti-inflammatory activity. In our test, compounds 1–3 were found not to reduce production of nitric oxide (NO) in RAW264.7 cells, and showed no cytotoxicity against RAW264.7 cells at the concentration of 40 µmol/L.
In the present study, three new aconitine-type C19-diterpenoid alkaloids novolunines A–C (1–3) were isolated from the roots of Aconitum novoluridum, and their structures were determined on the basis of phytochemical evidence. The plant Aconitum novoluridum has never been studied. Novolunines A (1) and B (2) are two diterpenoid alkaloids bearing conformational isomerism.
Optical rotations were measured in CH3Cl using a PerkinElmer, Inc. polarimeter (PerkinElmer, Inc., Waltham, U.S.A.) with a sodium lamp operating at 598 nm and 25 °C. IR spectra were obtained on a Thermo Fisher Nicolet 6700 spectrometer (Thermo Fisher Scientific, MA, U.S.A.). 1H- and 13C-NMR were measured in CDCl3, with tetramethylsilane (TMS) as internal standard, on a Bruker AV 400 NMR instrument. HR-ESI-MS spectra were measured by a Waters Acquity ultra performance liquid chromatography (UPLC)/Xevo G2-S QTof mass spectrometer. TLC silica gel GF254 plates and silica gel G and H (200–400 mesh) for column chromatography were purchased from Qingdao Ocean Chemical Plant (Qingdao, People’s Republic of China). The TLC plates were visualized under a UV lamp at 254 nm or by spraying the Dragendorff’s reagent or by iodine.
Plant MaterialThe roots of Aconitum novoluridum were collected in Basu County, Tibet Autonomous Region, People’s Republic of China, in July 2019. The plant was authenticated by Professor Liangke Song at the School of Life Science and Engineering, Southwest Jiaotong University, Sichuan Province, People’s Republic of China. A voucher specimen (A-N-2019714-2) has been deposited in Southwest Jiaotong University, Sichuan Province, People’s Republic of China.
Extraction and IsolationThe air-dried powder (16.0 kg) of Aconitum novoluridum was extracted with 95% EtOH three times at room temperature, with each socking process lasting five days. The combined filtrate was evaporated under reduced pressure to yield ethanol extract. The extract was suspended in H2O (4 L) and was adjusted to pH 3.0 with 10% HCl solution, followed by extracting with petroleum ether (4 × 3 L) and ethyl acetate (4 × 3 L). Then aqueous layer was alkalized with 28% aqueous ammonia solution to pH 10.0 and extracted with CH2Cl2 (6 × 3 L). The CH2Cl2 extract was combined and concentrated to give the total crude alkaloids (29.6 g). The crude alkaloids (29.6 g) were chromatographed over silica gel column eluting with CH2Cl2–MeOH (200 : 1→0 : 1) gradient system to give fractions A (2.8 g), B (3.7 g), C (2.9 g), D (2.1 g), E (3.9 g), and F (4.0 g). Fraction A (2.8 g) was chromatographed on a silica gel column eluting with CH2Cl2–MeOH (200 : 1→80 : 1) to afford compounds 4 (114 mg), 8 (25 mg), and two subfractions A1 and A2. Compounds 3 (29 mg) and 5 (25 mg) were isolated from subfraction A1 with petroleum ether–acetone (20 : 1→10 : 1). The subfraction A2 was further purified by RP-18 silica gel column with MeOH–H2O (40 : 60→65 : 35) to obtain compounds 1 (36 mg) and 2 (25 mg). Fraction B (3.7 g) was further chromatographed on a silica gel column eluted with petroleum ether–ethyl acetate (80 : 1→2 : 1) to give compounds 14 (33 mg), 9 (26 mg), 6 (13 mg), 7 (15 mg), 10 (17 mg), 11 (56 mg), 12 (12 mg), and 13 (43 mg). Fraction D (2.1 g) was chromatographed on a silica gel column and eluted with petroleum ether–acetone (15 : 1→2 : 1) to yield two subfractions (D1 and D2). D1 was further chromatographed on a silica gel column eluted with petroleum ether–ethyl acetate (15 : 1→5 : 1) to provide compounds 15 (14 mg) and 16 (20 mg). D2 was subjected to RP-18 silica gel column chromatography eluted with MeOH–H2O (30 : 70→55 : 45) to give compounds 17 (20 mg) and 18 (23 mg).
Novolunine A (1): White amorphous powder; [α]D25 + 16.3 (c = 0.1, CHCl3); IR (KBr) cm−1: 3397, 2964, 1710, 1609, 1592, 1263, 1204, 1095, 801; 1H-NMR (400 MHz, CDCl3) and 13C-NMR (100 MHz, CDCl3): see Table 1; HR-ESI-MS: [M + H]+ 670.3483, Calcd for C39H48N3O7+, 670.3487.
Novolunine B (2): White amorphous powder; [α]D25 + 3.5 (c = 0.2, CHCl3); IR (KBr) cm−1: 3417, 2924, 1698, 1608, 1292, 1091; 1H-NMR (400 MHz, CDCl3) and 13C-NMR (100 MHz, CDCl3): see Table 1; HR-ESI-MS: [M + H]+ 698.3794, Calcd for C41H52N3O7+, 698.3800.
Novolunine C (3): White amorphous powder; [α]D25−5.0 (c = 0.1, CHCl3); IR (KBr) cm−1: 3321, 2928, 1689, 1605, 1261, 1088; 1H-NMR (400 MHz, CDCl3) and 13C-NMR (100 MHz, CDCl3): see Table 1; HR-ESI-MS: [M + H]+ 553.2905, Calcd for C31H41N2O7+, 553.2908.
In Vitro Acetylcholinesterase Inhibition AssayAChE from Electrophorus electricus (electric eel), 5,5′-dithiobis-2-nitrobenzoic acid (Ellman’s reagent, DTNB), and acetylthiocholine chloride (ATC) were purchased from Macklin. The inhibitory activity of tested compounds 1–3 against AChE was assessed by Ellman’s method. Tested compounds were dissolved in DMSO to 50 µM. All the assays were under 0.2 M NaH2PO4/Na2HPO4 buffer (pH 6.7), using a spectraMax absorbance reader instrument. Enzyme solutions were prepared by 2.5 mg (0.5 U/mL) AChE in pH 8.0 buffer (1 mL). The assay medium contained phosphate buffer (pH 6.7, 140 µL), tested compounds (I = 50 µM, 10 µL), 0.5 U/mL of enzyme (10 µL). After 20 min of incubation time, DTNB (0.75 mM, 10 µL) and acetylthiocholine chloride (1.5 mM, 10 µL) were added for incubation for another 20 min. The inhibitory activity was determined by measuring the increase in absorbance at 405 nm. Each concentration was assayed in triplicate. Enzyme Inhibitory activity (%) = [1 − (A sample-blank/A control-blank)] × 100%.
Assay for Inhibition Ability Against Lipopolysaccharide (LPS)-Induced NO Production and Cytotoxicity TestingThe inhibitory activity of the isolated compounds toward NO production by RAW264.7 cells was determined using Griess reagent as previously described.32) RAW264.7 cells (ATC C) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS) in a humidified 5% CO2/95% air atmosphere at 37 °C. RAW264.7 cells were seeded in 96-well plates at 5 × 104 cells/well for NO production. The plates were pretreated with compounds 1–3 (40 µM/L) for 30 min and then incubated for 24 h with or without 1 µg/mL of LPS (Escherichia coli, serotype O111: B4, Sigma, U.S.A.). The nitrite concentration in the culture supernatant was measured by the Griess reaction. An amount of 100 µL of each culture supernatant was mixed with the same volume of Griess reagent (1% sulfanilamide in 5% phosphoric acid, and 0.1% naphthylethylenediamine dihydrocholoride in water) and the absorbance of the mixture at 550 nm was measured using a microplate reader. Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich). The absorbance at 540 nm was read using a microplate reader (POLARstar).
This work was supported by Grants of NSFC (31870329 and 82073734).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.