Regular Articles
Synthesis of Chloro-Substituted 6H-Dibenzo[b,d]pyran-6-one Natural Products, Graphislactone G, and Palmariols A and B
2021 年 69 巻 8 号 p. 781-788
詳細
2021 年 69 巻 8 号 p. 781-788
A palladium-mediated intramolecular aryl–aryl coupling reaction was applied to the total synthesis of the bioactive natural products, graphislactone G (1), and palmariols A (2) and B (3), which possess an unusual chloro-subsutituent on the 6H-dibenzo[b,d]pyran-6-one skeleton. Based on the transition state model of the coupling reaction, the mechanistic aspect for the regioselectivity of the aryl–aryl coupling reaction is also discussed.
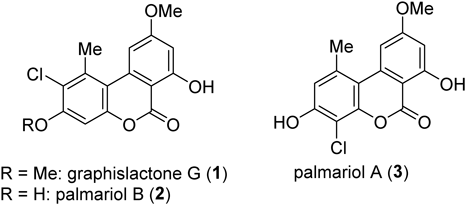
A variety of natural products, which possess the 6H-dibenzo[b,d]pyran-6-one core has attracted our interest because of their unique biological activities.1–18) Among them, graphislactone G (1) was isolated from Cephalosporium acremonium IFB-E007 in 2005 and known to exhibit an inhibitory activity against the SW1116 cell line.19) In addition, palmariols A (2) and B (3) were found from Lachnum palmae in 2010,20) and their antimicrobial, antinematodal, and acetylcholinesterase inhibitory activities were investigated.21) The outstanding structural feature of compounds 1–3 is that these compounds commonly possess a chlorine atom on the 6H-dibenzo[b,d]pyran-6-one ring (Fig. 1). Natural products, which possess a halogen atom on their aromatic ring, have attracted much attention because of their interesting biological activities.22) Thus, the total syntheses of such compounds have also intrigued synthetic chemists as well.
We have reported several natural product syntheses using the Pd-mediated intramolecular biaryl coupling reaction23–31) with phenyl benzoate derivatives for forming the 6H-dibenzo[b,d]pyran-6-one ring system.32–40) Utilizing this transformation, we planned the efficient syntheses of 1, 2, and 3. In Chart 1, our simple synthetic plan is shown in which the sequential transformation of esterification between the corresponding benzoic acid and phenol, followed by the Pd-mediated coupling reaction is involved.
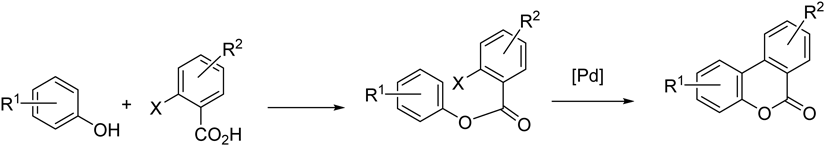
According to Chart 1, we commenced the synthesis of 141) by the preparation of the phenolic compound 5 involving the regioselective chlorination of 4 using N-chlorosuccinimide (NCS) (Chart 2). On the other hand, for the preparation of benzoic acid 9, 3,5-dimethoxyaniline (6) was subjected to the Sandmeyer reaction to substitute the amino group into the iodide to form 7. The following Vilsmeiyer reaction for introducing the formyl group was conducted to afford 8, then the Pinnick oxidation successfully afforded the carboxylic acid 9. After the conversion of 9 into the acid chloride, the condensation reaction with 5 produced the key intermediate 10 in a moderate yield accompanied by a small amount of the bis-ester 11, and unreacted 10 was also recovered. All efforts to improve the yield of 10 were unsuccessful in this step. The remaining hydroxy group of 10 was methylated, thus the coupling precursor 12 was prepared.

Using the obtained ester 12, we examined the intramolecular coupling reaction with the Pd reagent under several conditions. By this transformation, two regioisomerers, 13a and 13b, could be generated as the products. The chemical yields and the regioselectivities of 13a and 13b are listed in Table 1. When K2CO3 was employed as the base, and the bulkier phosphine ligand was used, a better regioselectivity was observed with 13a being the major product (runs 1–3). On the contrary, using Ag2CO3 as the base, only a low selectivity was observed (run 4).29,30) In order to validate the effect of the chlorine atom of 12, the previously reported results are shown in Table 1.42) The substrate 14, which possesses no chlorine atom on the aromatic ring, was subjected to the same conditions as runs 1–4 listed in Table 1. As shown in Table 1 (runs 5–8), both regioisomers 15a and 15b were produced with very low selectivity.
 |
This interesting regioselectivity could be explained by considering the transition state models.23,43–45) As illustrated in Fig. 2, model A presents the transition stage after the oxidative addition of Ar-I to Pd(0), and reacts at the ortho-position to the methyl group. Based on this model, the major product 13a is expected to be produced. On the other hand, in model B, the attractive interaction between the Pd atom and the ether oxygen must be considered.29,46) Due to the tight coordination, a severe steric hindrance is assumed around the chloro-substituent. This steric factor will make the transition state B significantly disfavored. Consequently, it would be reasonable that when using compound 12 as the substrate, 13a was mainly produced during the coupling reaction.
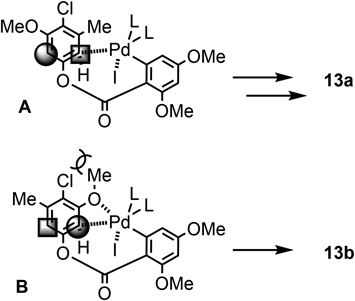
In a sharp contrast, when substrate 14 was used in a reaction similar to the above conditions, only a poor regioselectivity was observed, thus the coupling products 15a and 15b were obtained with the ratio of 1.5 : 1 to 0.6 : 1.42) Apparently, we recognized the effect of the chloro-substituent on the regioselectivity. In the case of 14, there is slight steric repulsion around the methoxy group like the transition state model B. As the result, the predominant regioselectivity would not be observed when using 14.
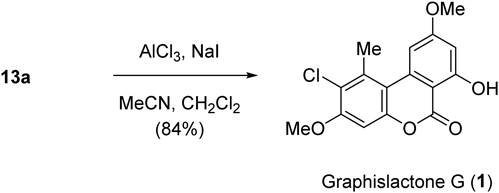
Finally, the selective demethylation reaction47) of the major coupling product 13a using Node’s protocol48) was successful to complete the synthesis of graphislactone G (1) (Chart 3).
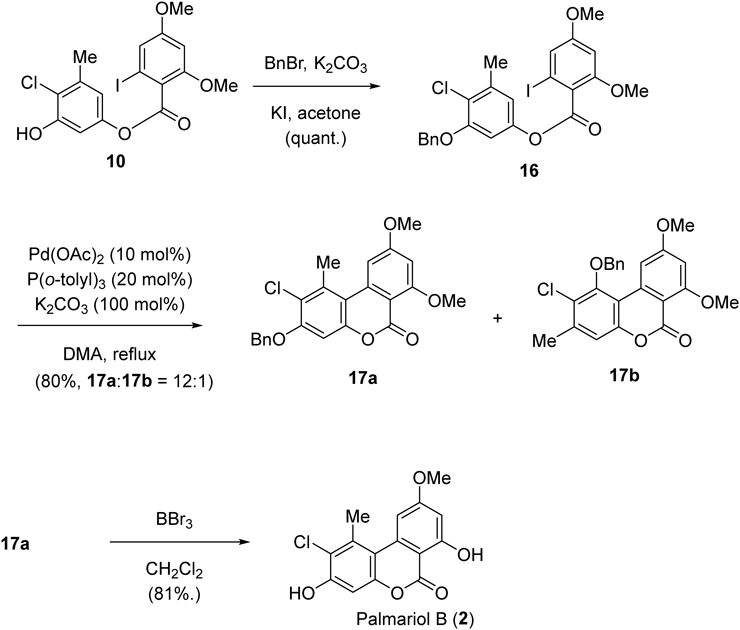
As an application of the above synthesis of graphislactone G, we next tried the synthesis of palmariol B (2) using a similar method (Chart 4). The key intermediate 10 was tentatively protected by the benzyl group on the phenolic hydroxy group. The quantitatively obtained 16 was subjected to the intramolecular coupling reaction under the same conditions as run 1 in Table 1. As expected, lactone compounds 17a and 17b were formed with a high regioselectivity (12 : 1). The major isomer 17a was reacted with boron tribromide for ether bond cleavage,47) thus the benzyl group was removed, and the selective demethylation simultaneously occurred. Therefore, the synthesis of palmariol B (2) was achieved.
For the synthesis of palmariol A (3), 5-methylcyclohexane-1,3-dione (18) was selected as the starting material, which was treated with chloramine-T to afford the chloride 19 (Chart 5). In the presence of p-toluenesulfonic acid, the reaction of 19 with benzyl alcohol generated the benzyl enol ether 20 which was converted to the silyl enol ether using lithium diisopropylamide (LDA) and trimethylsilyl chloride (TMSCl), followed by oxidation with iodine to form the phenol 21. The esterification between 21 and 9 successfully provided the coupling precursor 22. The palladium-mediated coupling reaction was conducted with the combination of Pd(OAc)2, PPh3, and Ag2CO3, forming the lactone 23 in a good yield. The final deprotection using BBr3 was also successful, thus the total synthesis of palmariol A (3) was completed.

We achieved the total synthesis of three natural products, graphislactone G and palmariols A and B, which possess a chloro-substituent on the aromatic ring. In the synthesis, the palladium-mediated intramolecular aryl–aryl coupling reaction was found to be very useful for the synthesis of these types of natural products.
Melting points were measured using a Yanagimoto micro-melting point hot plate apparatus and are uncorrected. The IR spectra were recorded using a JASCO FT/IR-305 or Shimadzu FTIR-8400 spectrophotometer. The NMR spectra were obtained using a Varian MERCURY (300 MHz), JEOL α-400 or JNX-ECX500 instrument. The chemical shifts are given in δ ppm with a solvent peak as the internal standard. The elemental analyses were performed using a Yanaco MT-5, Elementar Vario MICRO Cube or Thermo Fisher Scientific Flash EA 1112 analyzer. The MS were measured in the positive ion mode using a VG 70-SE or JEOL JMS-AX505HAD instrument. Silica gel column chromatography was carried out using Merck 9385 Kieselgel 60, Wakogel C-200 or C-400. The reaction solvents were used after purification by a standard method.
4-Chloro-1,3-dihydroxy-5-methylbenzene (5)49,50)To a solution of 1,3-dihydroxy-5-methylbenzene (4) (1.00 g, 8.06 mmol) in CHCl3 (80 mL) was added N-chlorosuccinimide (1.28 g, 9.67 mmol). The mixture was stirred at room temperature (r.t.) for 3 h, poured into water, and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4 and concentrated. The solid residue was purified by recrystallization from CH2Cl2 to give colorless needles of 5 (0.92 g, 72%), mp 132–133.5 °C (CH2Cl2) [lit.49) mp 137–139 °C]. IR (KBr) cm−1: 3320, 1615, 1600, 1440, 1340, 1270, 1160, 990. 1H-NMR (300 MHz, CDCl3) δ: 2.31 (3H, d, Me), 4.81 (1H, br s, 5-OH), 5.59 (1H, s, 1-OH), 6.32–6.33 (1H, m, H-4), 6.39–6.40 (1H, m, H-6).
3,5-Dimethoxyiodobenzene (7)51–53)To a mixture of 3,5-dimethoxyaniline (6) (0.102 g, 0.65 mmol) and a conc. HCl aqueous solution (0.2 mL) at 0 °C was added a solution of NaNO2 (0.053 g, 0.72 mmol) in water (3 mL). Ice (5 g) was added to the mixture which was stirred for 20 min at ambient temperature. A solution of KI (37.0 g, 223 mmol) in water (30 mL) was dropwise added to the mixture, and the mixture was then stirred at r.t. for 1 d. After extractive work-up with CH2Cl2, the organic layer was successively washed with a 5% NaHCO3, 10% Na2S2O3 aqueous solution, and brine, then dried over anhydrous MgSO4 and concentrated. Purification by silica gel column chromatography (CH2Cl2-hexane = 1 : 2) gave colorless prisms of 7 (0.121 g, 69%), mp 67 °C (CH2Cl2-hexane) [lit.51) 74–75 °C]. IR (KBr) cm−1: 2930, 1575, 1468, 1424, 1253, 1161, 984. 1H-NMR (300 MHz, CDCl3) δ: 3.76 (6H, s, ArOMe-3, ArOMe-5), 6.40 (1H, t, J = 2.4 Hz, H-4), 6.85 (2H, d, J = 2.1 Hz, H-2, H-6). Beilstein test: positive.
2-Iodo-4,6-dimethoxybenzaldehyde (8)54)To a solution of 7 (26.6 g, 101 mmol) in N,N-dimethylformamide (DMF) (20 mL) was dropwise added POCl3 (56 mL, 60 mmol). After the mixture was stirred at r.t. for 45 min, it was heated at reflux for 3 h. The mixture was poured into ice water to form a precipitate which was collected on a filter. An almost pure material of 8 (28.6 g, 97%) was obtained. For an analytical sample, recrystallization from CH2Cl2-hexane was carried out to give colorless needles, mp 75–76 °C [lit.54) 80–80.5 °C]. IR (KBr) cm−1: 2884, 1671 (CO), 1591, 1553, 1452, 1407, 1285, 1188, 1170, 1052, 933. 1H-NMR (300 MHz, CDCl3) δ: 3.86 (3H, s, 4-ArOMe), 3.89 (3H, s, 6-ArOMe), 6.47 (1H, d, J = 2.1 Hz, H-5), 7.12 (1H, d, J = 2.1 Hz, H-3), 10.13 (1H, s, CHO).
2,4-Dimethoxy-6-iodobenzoic Acid (9)55)To a mixture of 8 (491 mg, 1.68 mmol), a 31% H2O2 (0.18 mL, 1.85 mmol) aqueous solution, NaH2PO4·2H2O (289 mg, 1.85 mmol), MeCN (5 mL), and H2O (1 mL) was added a solution of 80% NaClO2 (209 mg, 1.85 mmol) in H2O (1 mL). The mixture was stirred for 3 h at r.t., then quenched with NaHSO3. After an extractive work-up with ether, the organic layer was washed with brine, dried over MgSO4, and concentrated to give a colorless solid of 9 (474 mg, 92%). An analytical sample was obtained by recrystallization from acetone–hexane, mp 145–147 °C. IR (KBr) cm−1: 3300 (OH), 1729 (CO). 1H-NMR (300 MHz, CDCl3) δ: 3.82 (3H, s, 2-OCH3), 3.85 (3H, s, 4-OCH3), 6.46 (1H, d, J = 2.1 Hz, H-3), 7.00 (1H, d, J = 2.1 Hz, H-5). Anal. Calcd for C9H9IO4: C, 35.09; H, 2.94. Found: C, 35.00; H, 2.88.
4-Chloro-3-hydroxy-5-methylphenyl 2-Iodo-4,6-dimethoxybenzoate (10)A mixture of SOCl2 (0.11 mL), 9 (102 mg, 0.33 mmol), and DMF (1 drops) was stirred for 30 min at 76 °C. After the volatile materials were removed under reduced pressure, the resulting residue was dissolved in dry CH2Cl2 (15 mL). Under cooling in an ice bath, 5 (38 mg, 0.24 mmol) was added to the solution. The mixture was stirred for 1 d at 0 °C and then poured into water. After acidification with a 10% HCl aqueous solution, the mixture was extracted with CHCl3. The organic layer was washed with brine, dried over MgSO4, and concentrated. The resulting residue was subjected to silica gel column chromatography (CH2Cl2) to give an amorphous solid of 10 (44 mg, 41%) and a trace amount of 11.
10: IR (KBr) cm−1: 3470, 3400, 3100, 3000, 2945, 2840, 1745, 1725, 1600, 1560, 1470, 1410, 1340, 1310, 1270, 1250, 1220, 1150, 1130, 1090, 1050, 1030, 975, 940, 890, 850, 820, 790, 770, 680. 1H-NMR (300 MHz, CDCl3) δ: 2.40 (3H, s, Me), 3.82 (3H, s, 4-OMe), 3.85 (3H, s, 6-OMe), 5.75 (1H, br s, OH), 6.48 (1H, d, J = 2.1 Hz, H-5), 6.79 (1H, dd, J = 2.7 Hz, 0.6 Hz, H-6′), 6.82 (1H, d, J = 2.7 Hz, H-2′), 6.96 (1H, d, J = 2.1 Hz, H-3). 13C-NMR (75 MHz, CDCl3) δ: 20.3 (Me), 55.8 (OMe), 56.1 (OMe), 92. 8, 98.9, 107.3, 115.3, 115.7, 118.0, 122.0, 137.5, 149.5, 152.0, 158.2, 161.9, 165.9. MS (FAB, positive ion mode) m/z: 449; 451 [M + 1]+.
4-Chloro-3-methoxy-5-methylphenyl 2-Iodo-4,6-dimethoxybenzoate (12)A mixture of 10 (250 mg, 0.56 mmol), K2CO3 (116 mg, 0.84 mmol), and tetrahydrofuran (THF) (12 mL) was stirred for 30 min at r.t.. To the mixture, methyl iodide (MeI) (0.17 mL, 2.79 mmol) was added, then heated at reflux for 16 h. The mixture was poured into water and extracted with ether. The organic layer was washed with brine, dried over MgSO4, and concentrated. The resulting solid was purified by recrystallization from ether to give colorless needles of 12 (228 mg, 88%), mp 129.5–130.5 °C (Et2O). IR (KBr) cm−1: 3080, 3020, 2960, 2940, 2840, 1755, 1600, 1560, 1440, 1410, 1325, 1300, 1250, 1220, 1150, 1100, 1035, 890, 815, 680. 1H-NMR (300 MHz, CDCl3) δ: 2.41 (3H, s, Me), 3.83 (3H, s, 4-OMe), 3.86 (3H, s, 6-OMe), 3.91 (3H, s, 3′-OMe), 6.50 (1H, d, J = 2.1 Hz, H-5), 6.76 (1H, d, J = 2.7 Hz, H-2′), 6.84 (1H, dd, J = 2.7 Hz, 0.6 Hz, H-6′), 6.97 (1H, d, J = 2.1 Hz, H-3). 13C-NMR (75 MHz, CDCl3) δ: 20.4 (Me), 55.7 (OMe), 56.1 (OMe), 56.3 (3-C-OMe), 92.8, 99.0, 103.5, 115.3, 115.6, 119.9, 122.1, 138.3, 149.3, 155.5, 158.1, 161.9, 165.7. Anal. Calcd for C17H16ClIO5: C, 44.13; H, 3.49. Found: C, 43.97; H, 3.52.
Typical Procedure for Table 1 (Run 1)Under an argon atmosphere, a mixture of 12 (50.0 mg, 0.11 mmol), Pd(OAc)2 (2.4 mg, 0.01 mmol), K2CO3 (14.9 mg, 0.11 mmol), P(o-tolyl)3 (6.6 mg, 0.02 mmol), and N,N-dimethylacetamide (DMA) (1.5 mL) was stirred for 30 min with heating at 190 °C. The mixture was diluted with AcOEt, then the undissolved materials were removed by filtration. The filtrate was poured into water and the mixture was extracted with AcOEt. The organic layer was washed with brine, dried over MgSO4, and concentrated to give a residue which was subjected to silica gel column chromatography (AcOEt : hexane = 1 : 2). A mixture of 13a and 13b (22.5 mg, 63%) was obtained with >20 : 1 ratio. For the analytical samples, further purification was carried out.
2-Chloro-3,7,9-trimethoxy-1-methyl-6H-dibenzo[b,d]pyran-6-one (13a)Colorless needles, mp 215.5–216.5 °C (Et2O-CH2Cl2). IR (KBr) cm−1: 3000, 2945, 2380, 1745, 1710, 1610, 1580, 1450, 1420, 1390, 1340, 1280, 1250, 1210, 1200, 1170, 1145, 1100, 1060, 1040, 1000, 960, 820, 730, 670. 1H-NMR (300 MHz, CDCl3) δ: 2.85 (3H, s, Me), 3.94 (3H, s, 9-OMe), 3.96 (3H, s, 3-OMe), 4.02 (3H, s, 7-OMe), 6.55 (1H, d, J = 2.1, H-8), 6.78 (1H, s, H-4), 7.13 (1H, d, J = 2.1 Hz, H-10). 13C-NMR (75 MHz, CDCl3) δ: 21.3 (Me), 55.6 (OMe), 56.4 (OMe × 2), 97.3 (Ar-CH), 98.3 (Ar-CH), 103.2 (Ar-CH), 103.8 (C-6a), 112.2 (C-10b), 120.5 (C-2), 134.8 (C-1), 139.6 (C-10a), 151.5 (C-7), 155.9 (C-4a), 157.6 (C-3), 163.7 (C-6), 164.6 (C-9). Anal. Calcd for C17H16O5: C, 60.99; H, 4.52. Found: C, 60.84; H, 4.75.
2-Chloro-1,7,9-trimethoxy-3-methyl-6H-dibenzo[b,d]pyran-6-one (13b)Colorless needles, mp 203.5–205.5 °C (Et2O-CH2Cl2). IR (KBr) cm−1: 3000, 2950, 2850, 1730, 1600, 1580, 1460, 1430, 1380, 1330, 1250, 1215, 1170, 1100, 1070, 1020, 1000, 940, 880, 845, 690. 1H-NMR (300 MHz, CDCl3) δ: 2.45 (3H, s, Me), 3.88 (3H, s, 1-OMe), 3.97 (3H, s, 9-OMe), 4.01 (3H, s, 7-OMe), 6.62 (1H, d, J = 2.1 Hz, H-8), 7.04 (1H, s, H-4), 8.20 (1H, d, J = 2.1 Hz, H-10). 13C-NMR (75 MHz, CDCl3) δ: 20.7 (Me), 55.6 (OMe), 56.4 (OMe), 60.2 (OMe), 99.5 (Ar-CH), 101.4 (Ar-CH), 103.7, 111.0, 115.0 (Ar-CH), 124.8, 137.9, 139.7, 150.5, 154.0, 157.4, 163.7, 165.7. Anal. Calcd for C17H16O5·3/4H2O: C, 58.63; H, 4.48. Found: C, 58.89; H, 4.47. MS (FAB, positive ion mode) m/z: 335, 337 [M + 1]+.
Graphislactone G (2-Chloro-7-hydroxy-3,9-dimethoxy-1-methyl-6H-dibenzo[b,d]pyran-6-one) (1)19,56)To a mixture of 13a (42.0 mg, 0.13 mmol), dry CH3CN (1.5 mL), and dry CH2Cl2 (2 mL) at 0 °C was successively added AlCl3 (83.7 mg, 0.63 mmol) and NaI (65.8 mg, 0.44 mmol). The mixture was warmed to r.t. and stirred for 25 min, then quenched by adding a 5% Na2S2O3 aqueous solution. After extractive work-up with CH2Cl2, the organic layer was washed with brine, dried over MgSO4, and concentrated to give a residue. Recrystallization from Et2O–CH2Cl2 left colorless needles of 1 (33.7 mg, 84%), mp 250.5–252.5 °C (Et2O–CH2Cl2) [lit.56) mp 243–245 °C]. IR (KBr) cm−1: 3165, 3082, 2945, 2850, 1670, 1630, 1596, 1580, 1460, 1420, 1390, 1350, 1280, 1235, 1205, 1160, 840, 770. 1H-NMR (300 MHz, CDCl3) δ: 2.89 (3H, s, Me), 3.92 (3H, s, 9-OMe), 3.97 (3H, s, 3-OMe), 6.57 (1H, d, J = 2.1, H-8), 6.82 (1H, s, H-4), 7.20 (1H, d, J = 2.1 Hz, H-10), 11.79 (1H, br s, OH, exchangeable with D2O). 13C-NMR (75 MHz, CDCl3) δ: 21.2 (Me), 55.8 (C-9-OMe), 56.5 (C-3-OMe), 98.9 (C-4), 99.4 (C-8), 99.6 (C-6a), 105.5 (C-10), 112.1 (C-10b), 121.5 (C-2), 135.7 (C-1), 137.1 (C-10a), 150.8 (C-4a), 156.0 (C-3), 165.0 (C-6), 165.1 (C-7), 166.3 (C-9). Anal. Calcd for C16H13ClO5: C, 59.92; H, 4.09. Found: C, 59.74; H, 4.19.
3-Benzyloxy-4-chloro-5-methylphenyl 2-Iodo-4,6-dimethoxybenzoate (16)To a solution of 10 (50 mg, 0.11 mmol) in acetone (3 mL) were added K2CO3 (35 mg, 0.25 mmol) and KI (39 mg, 0.23 mmol). BnBr (0.026 mL, 0.22 mmol) was then dropwise added to the mixture and stirred for 3 d at r.t. The mixture was poured into water and extracted with AcOEt. The organic layer was washed with brine, dried over MgSO4, and concentrated. The resulting residue was purified by silica gel column chromatography to give 16 (60 mg, quant.) in a pure form, colorless needles, mp 107.4–108.4 °C (Et2O–CH2Cl2). IR (KBr) cm−1: 1755, 1600, 1560, 1450, 1330, 1310, 1250, 1220, 1155, 1140, 1030, 860, 740. 1H-NMR (400 MHz, CDCl3) δ: 2.42 (3H, s, Me), 3.82 (3H, s, 4-OMe), 3.84 (3H, s, 6-OMe), 5.16 (2H, s, ArOCH2), 6.48 (1H, d, J = 2.2 Hz, H-5), 6.81 (1H, d, J = 2.6 Hz, H-2′), 6.85 (1H, d, J = 2.6 Hz, H-6′), 6.95 (1H, d, J = 2.2 Hz, H-3), 7.30–7.48 (5H, m, Ph-H). 13C-NMR (100 MHz, CDCl3) δ: 21.6 (Me), 56.8 (OMe), 57.2 (OMe), 72.1, 94.0, 100.1, 106.5, 116.4, 117.1, 121.8, 123.2, 128.2, 129.1, 129.7, 137.4, 139.4, 150.1, 155.7, 159.2, 163.0, 166.7. Anal. Calcd for C7H8O3: C, 51.27; H, 3.74. Found: C, 50.94; H, 3.64.
3-Benzyloxy-2-chloro-7,9-dimethoxy-1-methyl-6H-dibenzo[b,d]pyran-6-one (17a) and 1-Benzyloxy-2-chloro-7,9-dimethoxy-3-methyl-6H-dibenzo[b,d]pyran-6-one (17b)According to the procedure listed in Table 1, the reaction of 16 (29.6 mg, 0.055 mmol) afforded a mixture of 17a and 17b (80% yield with 12 : 1 ratio).
17a: Colorless needles, mp 145.3–146.3 °C (Et2O–CH2Cl2). IR (KBr) cm−1: 2950, 1745, 1710, 1605, 1580, 1460, 1420, 1380, 1345, 1280, 1250, 1210, 1190, 1165, 1150, 1100, 1070, 1030, 1000, 820, 720, 690. 1H-NMR (400 MHz, CDCl3) δ: 2.87 (3H, s, Me), 3.96 (3H, s, 9-OMe), 4.01 (3H, s, 7-OMe), 5.21 (2H, s, 3-ArOCH2), 6.54 (1H, d, J = 2.2 Hz, H-8), 6.80 (1H, s, H-4), 7.13 (1H, d, J = 2.2 Hz, H-10), 7.32–7.48 (5H, m, Ph-H). 13C-NMR (100 MHz, CDCl3) δ: 21.5, 55.7, 56.5, 70.9, 97.6, 99.9, 103.4, 104.1, 112.6, 121.3, 127.1, 128.3, 128.8, 135.0, 135.8, 139.8, 151.4, 155.1, 157. 7, 163.9, 164.7. Anal. Calcd for C23H19O5: C, 67.24; H, 4.66. Found: C, 67.06; H, 4.56.
17b: Colorless needles, 1H-NMR (500 MHz, CDCl3) δ: 2.48 (3H, s, Me), 3.42 (3H, s, 1-OMe), 3.99 (3H, s, 9-OMe), 4.97 (2H, s, ArOCH2), 6.57 (1H, d, J = 2.0 Hz, H-8), 7.08 (1H, s, H-4), 7.36–7.56 (5H, m Ph-H), 8.14 (1H, d, J = 2.0 Hz, H-10). 13C-NMR (125 MHz, CDCl3) δ: 20.9, 55.4, 56.5, 74.6, 100.2, 101.3, 103.8, 111.5, 115.2, 125.1, 128.0, 128.6, 128.7, 136.4, 138.0, 139.9, 150.7, 152.6, 157. 4, 163.6, 165.8. Anal. Calcd for C23H19O5: C, 67.24; H, 4.66. Found: C, 67.04; H, 4.56.
Palmariol B (2-Chloro-3,7-hydroxy-9-methoxy-1-methyl-6H-dibenzo[b,d]pyran-6-one) (2)20)Under N2, to a solution of 17a (101.2 mg, 0.246 mmol) in dry CH2Cl2 (4 mL) was added BBr3 (60 µL, 0.632 mmol) at 0 °C. The mixture was slowly warmed to rt and stirred for 1 h, then poured into water. After extractive work-up with AcOEt, the organic layer was washed with brine, dried over MgSO4, and concentrated to give a yellow solid. Recrystallization from hexane-acetone afforded colorless needles of 2 (61.2 mg, 81%) in a pure form, mp 282.9–283.1 °C. IR (KBr) cm−1: 3243, 1669, 1610, 1570, 1441, 1358, 1266, 1220, 1197, 1109, 984, 826, 668. 1H-NMR (400 MHz, CDCl3) δ: 2.89 (3H, s, Me), 3.92 (3H, s, ArOMe), 6.01 (1H, s, ArOH), 6.58 (1H, d, J = 2.1 Hz, ArH), 6.95 (1H, s, ArH), 7.18 (1H, d, J = 2.1 Hz, ArH), 11.80 (1H, s, ArOH). 13C-NMR (125 MHz, dimethyl sulfoxide (DMSO)-d6) δ: 166.2, 164.4, 164.1, 154.9, 150.3, 137.1, 135.7, 120.0, 110.5, 104.7, 102.1, 99.8, 99.1, 56.1, 21.1. Anal. Calcd for C15H11O5Cl: C, 58.74; H, 3.62. Found: C, 58.64; H, 3.45.
2-Chloro-5-methylcyclohexane-1,3-dione (19)57)To a mixture of 18 (5.09g, 40 mmol), THF (10 mL), and water (50 mL) was dropwise added a solution of chloramine-T (13.85 g, 50 mmol) in water (50 mL). The mixture was stirred overnight and the resulting precipitate was filtered off. After the solution was acidified with a 10% HCl aqueous solution, it was then extracted with ether. The organic layer was washed with brine, dried over MgSO4, and concentrated. Recrystallization from toluene afforded colorless prisms of 19 (4.95 g, 77%), mp 160.8–161.5 °C 1H-NMR (400 MHz, CDCl3) δ: 9.69 (1H, br s, enol OH) 2.70 (2H, m, 6-CH2), 2.48–2.10 (3H, m, 5-CH and 4-CH2), 1.08 (1H, d, Me).
3-Benzyloxy-2-chloro-5-methylcyclohex-2-enone (20)A mixture of 19 (3.21 g, 20 mmol), p-TsOH·H2O (190 mg, 1 mmol), BnOH (6.7 mL, 60 mmol), and benzene (150 mL) was heated overnight in an oil bath for azeotropic dehydration using a Dean–Stark apparatus. After neutralization with a sat. NaHCO3 aqueous solution, the mixture was extracted with CHCl3. The organic layer was washed with brine, dried over MgSO4, and concentrated. The resulting residue was purified by silica gel column chromatography (AcOEt : hexane = 1 : 4) to give colorless needles of 20 (3.59g, 72%). IR (KBr) cm−1: 1657, 1585, 1457, 1409, 1365, 1322, 1299, 1243, 1218, 1045, 977, 911, 893, 843, 757, 702, 642, 589, 453. 1H-NMR (400 MHz, CDCl3) δ: 7.43 (5H, m, Ph), 5.27 (1H, d, J = 12.8 Hz, CH2(Bn)), 5.24 (1H, d, J = 12.8 Hz, CH2(Bn)), 2.81 (1H, dd, J = 12.4, 2 Hz, 6-CH2), 2.60 (1H, dd, J = 13.6 Hz, J = 1.5 Hz, 4-CH2), 2.31 (1H, t, J = 12.4 Hz, 6-CH2), 2.30–2.20 (1H, m, 5-CH), 2.18 (1H, t, J = 13.6 Hz, 4-CH2), 1.08 (3H, d, J = 6.4 Hz, Me). 13C-NMR (100 MHz, CDCl3) δ: 191.2, 169.8, 135.5, 129.0, 128.6, 126.8, 112.5, 70.6, 44.8, 35.0, 28.1, 20.8.
3-Benzyloxy-2-chloro-5-methylphenol (21)A solution of iPr2NH (214 µL, 1.52 mmol) and hexamethylphosphoramide (HMPA) (6 mL) in THF (25 mL) under N2 was cooled at −78 °C. To the solution, nBuLi (1.57 M in hexane) (0.97 mL) was dropwise added, then TMSCl (1 mL) was added at −78 °C. A solution of 20 (382 mg, 1.52 mmol) in THF (10 mL) was dropwise added at −78 °C, then warmed to r.t. The whole mixture was heated under reflux for 1h. A solution of I2 (462 mg, 1.82 mmol) in THF (10 mL) was added to the mixture via a cannula, then heated overnight under reflux. After cooling, the reaction was quenched with a sat. Na2SO3 aqueous solution and the mixture was extracted with CHCl3. The organic layer was washed with brine, dried over MgSO4, and concentrated. The resulting residue was subjected to silica gel column chromatography (AcOEt : hexane = 1 : 8) to afford colorless needles of 21 (248.2 mg, 66%), mp 70.0–70.3 °C (hexane). IR (KBr) cm−1: 3499, 1587,1453, 1351, 1192, 1097, 810, 739, 701. 1H-NMR (400 MHz, CDCl3) δ: 7.47–7.33 (5H, m, Ph), 6.52 (1H, d, J = 1.4 Hz, Ph(C2-H)), 6.40 (1H, d, J = 1.4 Hz, Ph(C4-H)), 5.57 (1H, s, OH), 5.12 (2H, s, CH2-Ph), 2.28 (3H, s, Me). 13C-NMR (100 MHz, CDCl3) δ: 154.4, 152.2, 138.2, 136.6, 128.7, 128.1, 127.1, 109.5, 106.5, 106.3, 70.8, 21.8. Anal. Calcd for C14H13ClO2: C, 67.61; H, 5.27. Found: C, 67.31; H, 5.08.
3-Benzyloxy-2-chloro-5-methylphenyl 2-Iodo-4,6-dimethoxybenzoate (22)Under N2, a mixture of 21 (54.7 mg, 0.22 mmol), 9 (81.3 mg, 0.264 mmol), EDC (88.5 mg, 0.462 mmol), and N,N-dimethyl-4-aminopyridine (DMAP) (2.5 mg, 0.018 mmol) was dissolved in CH2Cl2 (3 mL) and then allowed to stand overnight. The mixture was poured into water and extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4, and concentrated. The resulting residue was subjected to silica gel column chromatography (AcOEt : hexane = 1 : 4) to afford colorless needles of 22 (76.9 mg, 71%), mp 168.0–168.2 °C. IR (KBr) cm−1: 1752, 1596, 1558, 1456, 1407, 1310, 1279,1235, 1213, 1186, 1157, 1088, 1060, 1030, 837, 751, 699. 1H-NMR (400 MHz, CDCl3) δ: 7.49–7.32 (5H, m, Ph), 6.99 (1H, d, J = 2.2 Hz, Ph’(C5-H)), 6.99 (1H, d, J = 1.0 Hz, Ph(C6-H)), 6.74 (1H, d, J = 1.0 Hz, Ph(C4-H)), 6.50 (1H, d, J = 2.2 Hz, Ph(C3-H)) 5.15 (2H, s, CH2-Ph), 3.86 (3H, s, OMe), 3.82 (3H, s, OMe), 2.36 (3H, s, Me). 13C-NMR (100 MHz, CDCl3) δ: 165.2, 162.1, 158.6, 155.2, 147.7, 137.6, 136.5, 128.6, 128.0, 127.1, 121.9, 116.5, 115.6, 113.7, 112.4, 99.0, 93.1, 71.1, 56.1, 55.8, 21.7. Anal. Calcd for C23H20ClIO5: C, 51.27; H, 3.74. Found: C, 51.43; H, 3.64.
3-Benzyloxy-4-chloro-7,9-dimethoxy-1-methyl-6H-dibenzo[b,d]pyran-6-one (23)Under N2, a mixture of 22 (54 mg, 0.10 mmol), Pd(OAc)2 (2.2 mg, 0.010 mmol), Ag2CO3 (27.6 mg, 0.10 mmol), PPh3 (5.2 mg, 0.02 mmol), and DMA (2 mL) was stirred for 1 h under reflux. The mixture was diluted with AcOEt, then the undissolved materials were removed by filtration. The filtrate was poured into water and the mixture was extracted with AcOEt. The organic layer was washed with brine, dried over MgSO4, and concentrated to give a residue which was subjected to silica gel column chromatography (AcOEt : hexane = 1 : 2). Colorless crystals of 23 (33.3 mg, 81%) were obtained, mp 240.3–240.7 °C. IR (KBr) cm−1: 1727, 1599, 1575, 1470, 1341, 1283, 1208, 1174, 1144, 1110, 1074, 1029, 967, 835, 814, 741, 698, 684. 1H-NMR (400 MHz, CDCl3) δ: 7.50–7.34 (5H, m, Ph), 7.23 (1H, d, J = 2.4 Hz, C8-H)), 6.77 (1H, s, C2-H), 6.56 (1H, d, J = 2.4 Hz, C10-H), 5.24 (2H, s, CH2-Ph), 4.01 (3H, s, OMe), 4.00 (3H, s, OMe), 2.80 (3H, s, Me). 13C-NMR (100 MHz, CDCl3) δ: 164.9, 164.1, 156.8, 154.6, 149.6, 140.1, 136.1. 134.7, 128.8, 128.3, 127.1, 123.3, 112.7, 108.8, 103.6, 103.0, 97.6, 71.0, 56.6, 55.6, 25.9. HRMS(EI) m/z, Calcd: 410.0921, Found: 410.0933. Anal. Calcd for C23H19ClO5: C, 67.24; H, 4.66. Found: C, 67.35; H, 4.52.
Palmariol A (4-Chloro-3,7-dihydroxy-9-methoxy-1-methyl-6H-dibenzo[b,d]pyran-6-one) (3)20)Under N2, to a solution of 22 (41 mg, 0.10 mmol) in dry CH2Cl2 (2 mL) was added BBr3 (1.0 M in CH2Cl2) (0.44 mL, 0.44 mmol) at 0 °C. The mixture was slowly warmed to r.t. and stirred 20 min, then poured into water. After extractive work-up with AcOEt, the organic layer was washed with brine, dried over MgSO4, and concentrated to give a yellow solid which was subjected to silica gel column chromatography (AcOEt : hexane = 1 : 1). Colorless needles of 3 (23 mg, 75%) were obtained, mp. 288.0–288.2 °C. IR (KBr) cm−1: 3410, 1671, 1606, 1569, 1337, 1273, 1228, 1192, 1162, 1129. 1H-NMR (400 MHz, CDCl3) δ: 11.8 (1H, s, OH), 7.27 (1H, d, J = 1.6 Hz, C8-H)), 6.90 (1H, s, C2-H), 6.59 (1H, d, J = 1.6 Hz, C10-H), 5.84 (1H, s, OH), 3.92 (3H, s, OMe), 2.80 (3H, s, Me). 1H-NMR (400 MHz, Acetone-d6) δ: 11.81 (1H, s, OH), 7.33 (1H, d, J = 2.2 Hz, C8-H), 6.98 (1H, s, C2-H), 6.63 (1H, d, J = 1.6 Hz, C10-H), 3.99 (3H, s, OMe), 2.84 (3H, s, Me). 1H-NMR (400 MHz, DMSO-d6) δ: 11.64 (1H, s, OH), 7.23 (1H, d, J = 1.2 Hz, C8-H), 6.98 (1H, s, C2-H), 6.67 (1H, d, J = 1.2 Hz, C10-H), 3.92 (3H, s, OMe), 2.73 (3H, s, Me). 13C-NMR (125 MHz, Acetone-d6) δ: 167.7, 165.9, 165.3, 155.0, 149.5, 138.7, 137.1, 117.9, 112.0, 105.0, 100.4, 56.4, 25.4. 13C-NMR (125 MHz, DMSO-d6) δ: 166.3, 164.0, 163.8, 154.4, 148.1, 137.4, 135.9, 116.9, 110.1, 105.7, 103.9, 99.7, 98.3, 55.9, 25.0. HRMS(EI) m/z, Calcd: 306.0295, Found: 306.0293. Anal. Calcd for C15H11ClO5: C, 58.74; H, 3.62. Found: C, 58.95; H, 3.45.
The authors wish to acknowledge Mr. Shumpei Saga and Mr. Yutaka Kadoshima for their technical support. Funding from THE HOKURIKU BANK Grant-in-Aid for Young Scientists is gratefully acknowledged.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.