Regular Articles
Photoinduced Atom Transfer Radical Addition Reaction of Olefins with α-Bromo Carbonyls
2021 年 69 巻 8 号 p. 796-801
詳細
2021 年 69 巻 8 号 p. 796-801
The irradiation of halogen-bonded complexes with light leads to the homolysis of carbon–halogen bonds and the formation of the corresponding carbon radical species. However, the only methodology reported for these halogen-bonding complexes is using CBr4 as the halogen-bond donor and its applicability is of great interest. In this study, the atom transfer radical addition (ATRA) reaction of olefins using bromomalonates as halogen-bonding donors was developed. Using 4-phenylpyridine as the halogen-bonding acceptor, the desired reaction proceeded well under external irradiation of 380 nm light to furnish the corresponding ATRA reaction product. The ATRA reaction was effective in generating the corresponding products for a variety of olefins. Furthermore, the ATRA reaction was applicable to bulky ketones, substrates, and malonate esters. The intermediates of the reaction were identified and a plausible reaction mechanism was proposed.
Atom transfer radical addition (ATRA) of halogenated compounds to alkenes is a powerful and highly atom-economical method to simultaneously form carbon–carbon bonds and carbon–halogen bonds.1) Almost 70 years ago, this reaction was pioneered by Kharasch et al.2,3) Some of the earliest efforts involved the development of the ATRA reaction as a useful tool for the double functionalization of olefins, which are abundant and commercially available feedstocks.4–6) However, some initiators classically employed in established ATRA reactions are stoichiometric amounts of organotin reagents,4) triethyl borane,5) or potentially explosive oxidants,6) and a high reaction temperature is also required. In recent years, transition metal (TM) catalysis,7,8) including visible-light-driven photoredox catalysis (PC),9,10) has opened up further possibilities for ATRA reactions (Chart 1a). Nevertheless, a suitable method is needed to generate radical intermediates under mild reaction conditions without expensive transition metal catalysts or toxic reagents.
In this regard, ATRA reactions using organo-photocatalysts have been extensively studied.11,12) Particularly, the use of halogen bonding (HB), which is a noncovalent interaction between electron-deficient halogen compounds and Lewis bases,13–15) in organo-photocatalysis has recently attracted considerable attention16) (Chart 1b). It does not require transition metals or harsh conditions and is a potential method to replace the conventional ATRA reaction because interactions with simple organic molecules are used as active species. For example, in 2017, Miyabe and colleagues demonstrated that the formation of Charge–Transfer (CT) complexes between iodine and tertiary amines through halogen bonding has the potential to induce ATRA reactions.17) Additionally, in 2019, Czekelius and colleagues revealed that halogen bonding induced the metal-free photomediated activation of perfluoroalkyl iodides using phosphines or phosphites.18) However, photocatalysis through halogen bonding employing a catalytic amount of amine has not been extensively developed yet. Moreover, the range of substrates used in these reactions has been often limited to perfluoroalkanes with the introduction of strong electron withdrawing substituents.
Recently, we have reported that in situ generated halogen-bonding complex enabled the ATRA reactions of olefins.19) The carbohalogenation of olefins with carbon tetrabromide generated a variety of products in moderate to good yields when irradiated at 450 nm. The products obtained from the addition of carbon tetrabromide can be valuable intermediates for the synthesis of nitrogen heterocycles, such as pyrazoles.20) However, the adducts are not versatile synthetic intermediates. Therefore, to discover further applications for this reaction, we focused on the versatility of the derived adducts and attempted to apply them to bromomalonic acid instead of carbon tetrabromide (Chart 1c).
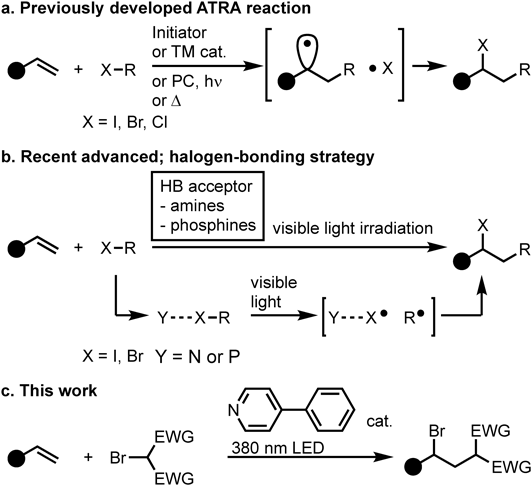
This study was initiated by examining the ATRA reaction between allylbenzene (1a) and dimethyl bromomalonate (2a) using the halogen-bonding approach (Table 1). Detailed optimization of the light source, and solvent is provided in Supplementary Materials. Herein, the most relevant observations are highlighted: upon purple light irradiation, a mixture of 1a and 2a with a catalytic amount of 4-phenylpyridine (4a) as a halogen-bonding acceptor in CH2Cl2 provided the corresponding ATRA product (3a) in 77% yield (entry 1).
 | |||
|---|---|---|---|
| Entry | Solvent | X (nm) | 3a (%)a) |
| 1 | CH2Cl2 | 380 | 77 |
| 2 | MeCN | 380 | 38 |
| 3 | DMF | 380 | 21 |
| 4 | THF | 380 | 0 |
| 5 | CH2Cl2 | 370 | 65 |
| 6 | CH2Cl2 | 390 | 62 |
| 7 | CH2Cl2 | 400 | 61 |
| 8b) | CH2Cl2 | 380 | 90 (78) |
a) Yields were determined through 1H-NMR analyses of the crude reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard. The number in parenthesis is the isolated yield. b) The reaction was performed using 175 mol% of 1a in CH2Cl2 (0.5 mL).
The use of a non-polar solvent such as CH2Cl2 was crucial to minimize the formation of undesired halogen-bonding complexes. In contrast, the use of polar and Lewis basic solvents such as MeCN, N,N-dimethylformamide (DMF), and tetrahydrofuran (THF) resulted in a low yield, owing to the formation of a halogen-bonding complex between 2a with the solvent (entries 2–4).21,22) Next, the wavelength of the light was evaluated. When the reaction was conducted by irradiation with light having wavelengths shorter or longer than 380 nm, the yield of 3a decreased slightly (entries 5–7). Therefore, 380 nm was selected as the optimal wavelength for this reaction. Finally, the loading of 1a was increased to 175 mol% and the volume of the solvent was reduced to 0.5 mL, which resulted in 78% yield of the desired 3a (entry 8).
The results of screening for halogen-bonding acceptors (4) are shown in Chart 2. Basically, the yield of employing pyridine with electron-withdrawing functional group (4b–4i) as a halogen-bonding acceptor tends to decrease compared with 4a. Since the lone pair of pyridine reacts with the σ-hole of the halogen in the formation of the halogen-bonding complex, we hypothesized that the electron-deficient pyridine would weaken this interaction and drop the reactivity. On the other hand, pyridines bearing electron-donating functional groups such as methoxy (4j), amino (4k, 4l and 4n) and alkyl groups (4n) show a moderate yield. Reaction with 2a in the presence of pyridines bearing a nucleophilic nitrogen moiety produced an insoluble salt. This byproduct has not been identified, but we believe that the formation of this salt, which is an adduct of bromide, caused the decrease in yield. We conclude that 4a is the best halogen-bonding acceptor for the present reaction system.
Yields were determined through 1H-NMR analyses of the crude reaction mixture using 1,1,2,2-tetrachloroethane as an internal standard. The number in parenthesis is the isolated yield.
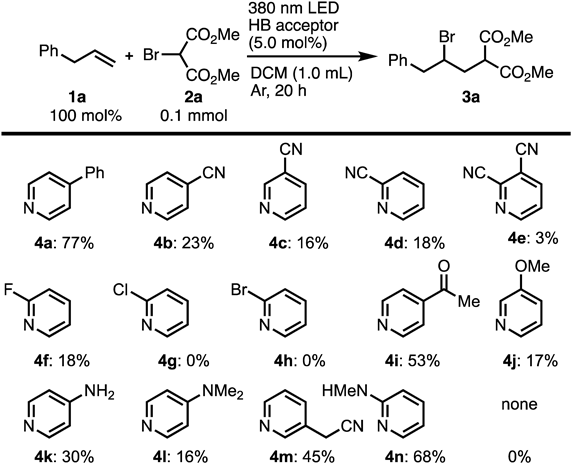
The scope of the photoinduced ATRA reaction was further explored using the optimized reaction conditions (Chart 3). 4-phenyl-1-butene (1b) with dimethyl bromomalonate (2a) produced the corresponding product in high yield, but the ATRA product of 1-(4-methoxyphenyl)-2-propene (1c) and dimethyl bromomalonate could only be achieved in low yield. The use of 1-(4-methoxyphenyl)-2-propene (1c) predominantly resulted in the alkene polymerization owing to the increased reactivity of the stabilized radical. Additionally, aliphatic olefins were subjected to the optimized ATRA reaction. The linear alkene-bearing decene (1d), dodecane (1e), and ester (1f) were all endured under these reaction conditions, providing the corresponding product in moderate to good yields. Interestingly, the primary alkyl bromides (1g and 1h) were adequate substrates and generated the ATRA adducts in moderate to good yields without any decomposition of the C–Br bond. Although the use of allyl alcohol (1i) and protected allyl alcohol (1j) as substrates resulted in low yields, the hexenol (1k) produced the product in high yield.

Next, the reactivity for various bromomalonates 2 with allylbenzene (1a) (Chart 4) was evaluated. When diethyl bromomalonate (2b) was used as the substrate, the corresponding ATRA adduct was obtained in moderate yield. However, when the 1,3-diketone (2c), in which the ester group was replaced by a carbonyl group, was used as a substrate, the corresponding product was obtained in low yield. Similarly, when a substrate containing an alkyl group, such as methyl (2d), n-propyl (2e), and cyanoethyl (2f), introduced into the active methylene moiety of bromomaloates was used, the reaction proceeded slowly. When bromomaloates containing benzyl (2g) or phenethyl (2h) groups were used as substrates, the reaction did not proceed.23)
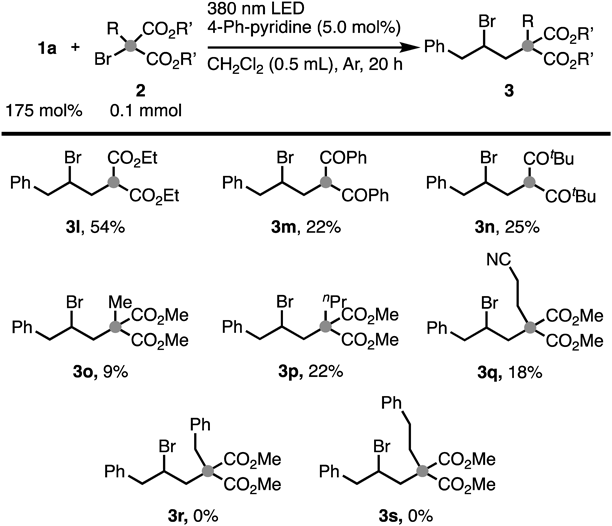
Further mechanistic investigations were also conducted (Chart 5). No reaction was observed between 1a and 2a using the standard conditions under an oxygen or air atmosphere (Chart 5a). The result indicated that the photoexcited active species was quenched by triplet oxygen species.24) Furthermore, the reaction was also investigated under dark conditions or heated to 60 °C under dark conditions, revealing that no adduct was obtained (Chart 5b). These results suggested that light is essential for this reaction and thermal activation of the molecules is not necessary. When the reaction was examined in the absence of catalyst, no product was obtained (Chart 5c), indicating that 4-Ph-pyridine acted as a catalyst.
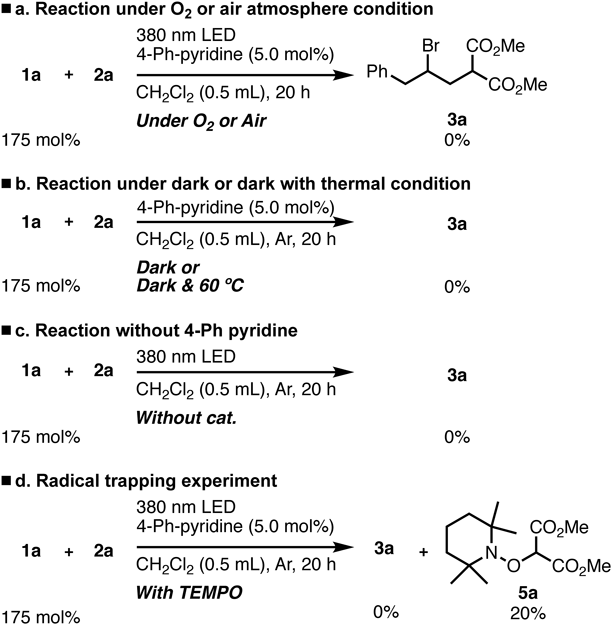
Moreover, no desired product was observed in the radical trapping experiment, whereas the use of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) under the optimized condition proceeded via the radical intermediate (Chart 5d). When TEMPO was used as a radical scavenger, the corresponding TEMPO adduct 5a was determined by NMR analysis.25)
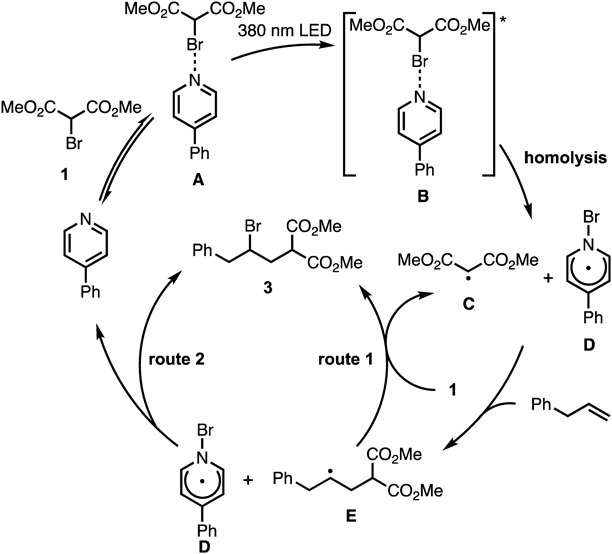
Based on the previously reported results,19) the possible reaction pathway for the generation of the malonate radical is shown in Chart 6. The reaction was initiated by photoexcitation of the halogen-bonding complex A to produce the excited state B. The photoexcitation of B probably led to the C–Br bond homolysis, generating the neutral radical complex C and the C-centered malonate radical D. Next, the resulting C-centered radical D reacted with the olefins to generate the radical intermediate E, which subsequently reacted with C to provide the desired ATRA product. Another possibility is a radical chain mechanism, in which the generated intermediate E reacted with bromomalonate to produce the ATRA adduct and regenerate D.
In summary, ATRA reactions of olefins with bromomalonate ester catalyzed by an in situ-formed halogen-bonding complex were investigated. The carbohalogenation of olefins with bromomalonate generated various products in moderate to good yields. The photocatalysts used in this study indicated that the halogen-bonding complex between pyridines and bromomalonate was a crucial intermediate in the subsequent charge separation processes that led to the formation of the C-centered radical.
The substrate activation through the halogen bonding from the CT complex is a novel activation method that can be used for challenging synthetic routes since halogen bond acceptors such as pyridine are relatively inexpensive and readily available. Further efforts are focused on extending this new methodology to other classes of compounds for photocatalysis, including alkyl halides with amines or phosphines.
Unless otherwise noted, all reactants or reagents including dry solvents were obtained from commercial suppliers and used as received.
Analytical TLC was carried out using 0.25 mm commercial silica gel plates (Merck silica gel 60 F254). Flash column chromatography was performed with Kanto silica gel 60N (Spherical, Neutral, 40–50 mm). Visualization of the developed chromatogram was performed by UV lamp (254 nm) and vanillin or basic potassium permanganate stain. NMR spectra were recorded on a JEOL ECA 500 spectrometer (500 MHz for 1H-NMR and 125 MHz for 13C-NMR), and are internally referenced to residual protio solvent signals or tetramethylsilane (TMS) (note: CDCl3 referenced at δ 7.26 and 77.0 ppm, respectively, TMS referenced at δ 0 and 0 ppm, respectively). Data for 1H-NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets, ddd = doublet of doublet of doublets, td = triplet of doublets), coupling constant (Hz), integration, and assignment. Data for 13C-NMR are reported in terms of chemical shifts (δ ppm). IR spectra were recorded on a PerkinElmer, Inc. Spectrum 100 Fourier transform (FT)IR spectrometer and are reported in terms of frequency of absorption (cm−1). High-resolution mass spectra (HRMS) were obtained on a JEOL JMS-T100TD and are reported as m/z (M + H+, relative intensity). Melting points were measured on a Yanagimoto micro melting point apparatus without correlation.
General Procedure for ATRAA Pyrex® test tube (12.5 cm × 1.6 cm) containing a mixture of alkene 1 (1.75 equivalent (equiv), 0.175 mmol), α-bromo carbonyl 2 (1.0 equiv, 0.1 mmol) and 4-phenylpyridine (0.05 equiv, 0.005 mmol) in dichloromethane (0.5 mL) was degassed via FPT cycling for three times and backfilled with Ar. The tube was placed approx. 0.5 cm from 3W 380 nm LED. The resulting solution was stirred at ambient temperature for 20 h. The residue was concentrated in vacuo. The resulting mixture was purified by flash column chromatography on silica gel to give product 3.
Characterization of ATRA Adduct 3Dimethyl 2-(2-Bromo-3-phenylpropyl)malonate (3a)3a (colorless oil); TLC (SiO2) Rf = 0.47 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.33 (t, J = 7.5 Hz, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.21 (d, J = 6.9 Hz, 2H), 4.24–4.18 (m, 1H), 3.87–3.82 (dd, J = 6.3, 4.0 Hz, 1H), 3.741 (s, 3H), 3.738 (s, 3H), 3.25–3.17 (m, 2H), 2.53 (td, J = 7.5, 3.7, 2.9 Hz, 1H), 2.24 (td, J = 6.9, 4.0, 4.0 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.3, 169.0, 137.6, 129.2, 128.5, 127.0, 54.0, 52.8, 52.7, 50.2, 45.7, 37.2; FTIR (attenuated total reflectance (ATR)) 3030, 2954, 2848, 1750, 1735, 1603, 1497, 1455, 1436, 1347, 1274, 1257, 1201, 1152, 1104, 1082, 1059, 1030, 1008, 975, 911, 845, 752, 701 cm−1; HRMS m/z direct analysis in real time (DART) Calcd for C14H18BrO4 (M + H)+ 329.0383. Found 329.0379.
Dimethyl 2-(2-Bromo-4-phenylbutyl)malonate (3b)3b (colorless oil); TLC (SiO2) Rf = 0.21 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.33 (t, J = 7.7 Hz, 2H), 7.22–7.19 (m, 3H), 3.97–3.92 (m, 1H), 3.85–3.83 (dd, J = 5.7, 4.6 Hz, 1H), 3.74 (s, 3H), 3.73 (s, 3H), 2.94–2.88 (m, 1H), 2.79–2.73 (m, 1H), 2.50 (td, J = 8.0, 6.9, 4.0, Hz, 1H), 2.33 (td, J = 6.3, 4.6, 4.0 Hz, 1H), 2.20–2.11 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.3, 169.1, 140.5, 128.50, 128.46, 126.2, 53.9, 52.8, 52.7, 50.2, 40.9, 37.9, 33.6; FTIR (ATR) 3063, 3028, 3004, 2954, 2857, 1949, 1750, 1734, 1603, 1584, 1497, 1454, 1435, 1342, 1260, 1242, 1201, 1151, 1069, 1016, 963, 906, 846, 749, 700, 655 cm−1; HRMS m/z (DART) Calcd for C15H20BrO4 (M + H)+ 343.0540. Found 343.0537.
Dimethyl 2-(2-Bromo-3-(4-methoxyphenyl)propyl)malonate (3c)3c (colorless oil); TLC (SiO2) Rf = 0.23 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.13 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.19–4.14 (m, 1H), 3.85 (dd, J = 6.9, 4.0 Hz, 1H), 3.80 (s, 3H), 3.74 (s, 6H), 3.19–3.11 (m, 2H), 2.79–2.71 (m, 1H), 2.52 (td, J = 8.0, 6.9, 4.0, Hz, 1H), 2.22 (td, J = 8.0, 6.9, 4.0 Hz, 1H), 2.18–2.11 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.3, 169.0, 158.5, 130.2, 129.7, 113.8, 55.2, 54.6, 52.8, 52.7, 50.2, 44.9, 37.1; FTIR (ATR) 3003, 2955, 2838, 1750, 1736, 1613, 1585, 1514, 1437, 1302, 1249, 1202, 1179, 1152, 1111, 1034, 975, 906, 832, 819, 759 cm−1; HRMS m/z (DART) Calcd for C15H20BrO5 (M + H)+ 359.0489. Found 359.0494.
Dimethyl 2-(2-Bromodecyl)malonate (3d)3d (colorless oil); TLC (SiO2) Rf = 0.64 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.00–3.95 (m, 1H), 3.84 (dd, J = 6.3, 4.0 Hz, 1H), 3.78 (s, 3H), 3.75 (s, 3H), 2.48 (td, J = 7.5, 4.6, 2.9 Hz, 1H), 2.26 (td, J = 8.0, 6.9, 4.0 Hz, 1H), 1.89–1.80 (m, 2H), 1.55–1.51 (m, 1H), 1.49–1.38 (m, 1H), 1.31–1.25 (m, 10H), 0.88 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.4, 169.2, 55.0, 52.8, 52.7, 50.2, 39.4, 37.9, 31.8, 29.4, 29.2, 28.9, 27.4, 22.6, 14.1; FTIR (ATR) 3299, 2127, 1640, 1452, 1111, 1016 cm−1; HRMS m/z (DART) Calcd for C15H27O4 (M-Br)+ 271.1904. Found 271.1909.
Dimethyl 2-(2-Bromododecyl)malonate (3e)3e (colorless oil); TLC (SiO2) Rf = 0.64 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.00–3.95 (m, 1H), 3.85 (dd, J = 6.3, 4.0 Hz, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 2.48 (td, J = 7.5, 4.6, 3.4 Hz, 1H), 2.26 (td, J = 8.6, 6.3, 4.6 Hz, 1H), 1.89–1.80 (m, 2H), 1.55–1.50 (m, 1H), 1.47–1.38 (m, 1H), 1.30–1.26 (m, 14H), 0.88 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.4, 169.2, 55.0, 52.8, 52.7, 50.2, 39.4, 37.9, 31.9, 29.54, 29.51, 29.4, 29.3, 28.9, 27.4, 22.7, 14.1; FTIR (ATR) 2924, 2854, 1743, 1463 cm−1; HRMS m/z (DART) Calcd for C17H32BrO4 (M + H)+ 379.1479. Found 379.1490.
Tetramethyl-1,1,5,5-(3-bromopentane)tetracarboxylate (3f)3f (colorless oil); TLC (SiO2) Rf = 0.24 (n-hexane : ethyl acetate = 5 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.04–3.99 (m, 1H), 3.80 (dd, J = 5.2, 4.6 Hz, 2H), 3.77 (s, 6H), 3.76 (s, 6H), 2.51 (td, J = 9.2, 5.7, 4.0 Hz, 2H), 2.33 (td, J = 9.2, 5.7, 4.6 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.1, 168.8, 52.9, 52.8, 51.1, 50.0, 37.9; FTIR (ATR) 3306, 2952, 2840, 2523, 2147, 1649, 1450, 1410, 1113, 1014 cm−1; HRMS m/z (DART) Calcd for C13H19O8 (M-Br)+ 303.1074. Found 303.1076.
Dimethyl 2-(2,4-Dibromobutyl)malonate (3g)3g (colorless oil); TLC (SiO2) Rf = 0.56 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.22–4.14 (m, 1H), 3.84 (dd, J = 5.7, 4.0 Hz, 1H), 3.79 (s, 3H), 3.77 (s, 3H), 3.62–3.54 (m, 2H), 2.51 (td, J = 6.9, 5.2, 2.9 Hz, 1H), 2.37–2.29 (m, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.1, 168.9, 52.9, 51.8, 50.0, 41.5, 37.6, 30.5; FTIR (ATR) 2955, 1736, 1436, 1270, 1249, 1207, 1156, 1019, 844 cm−1; HRMS m/z (DART) Calcd for C9H15Br2O4 (M + H)+ 344.9332. Found 344.9336.
Dimethyl 2-(2,5-Dibromopentyl)malonate (3h)3h (colorless oil); TLC (SiO2): Rf = 0.53 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.03–3.98 (m, 1H), 3.84 (dd, J = 6.3, 4.0 Hz, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 3.44 (t, J = 5.7 Hz, 2H), 2.49 (td, J = 8.0, 6.9, 3.4 Hz, 1H), 2.29 (td, J = 8.6, 6.3, 4.6 Hz, 1H), 2.22–2.14 (m, 1H), 2.09–1.96 (m, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.2, 169.1, 53.3, 52.9, 52.8, 50.1, 37.9, 37.7, 32.6, 30.4; FTIR (ATR) 2955, 1736, 1437, 1263, 1242, 1205, 1155, 1016 cm−1; HRMS m/z (DART) Calcd for C10H17Br2O4 (M + H)+ 358.9488. Found 358.9488.
Dimethyl 2-(2-Bromo-3-hydroxypropyl)malonate (3i)3i (colorless oil); TLC (SiO2) Rf = 0.30 (n-hexane : ethyl acetate = 2 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.19–4.14 (m, 1H), 3.87–3.80 (m, 3H), 3.78 (s, 3H), 3.77 (s, 3H), 2.52 (td, J = 9.2, 5.7, 4.0 Hz, 1H), 2.37 (td, J = 9.7, 5.2, 5.2 Hz, 1H), 2.27 (t, J = 6.9 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.3, 169.0, 66.9, 55.4, 52.92, 52.86, 49.6, 33.7; FTIR (ATR) 3479, 2956, 2051, 1730, 1436, 1342, 1258, 1201, 1152, 1114, 1068, 1022, 921, 844, 702 cm−1; HRMS m/z (DART) Calcd for C8H14BrO5 (M + H)+ 269.0019. Found 269.0025.
Dimethyl 2-(3-Acetyloxy-2-bromo-propyl)malonate (3j)3j (colorless oil); TLC (SiO2) Rf = 0.21 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.41–4.37 (m, 1H), 4.29–4.25 (m, 1H), 4.17–4.12 (m, 1H), 3.82–3.80 (m, 1H), 3.78 (s, 3H), 3.77 (s, 3H), 2.57 (td, J = 6.9, 3.4, 2.9 Hz, 1H), 2.26 (td, J = 8.0, 4.0, 4.0 Hz, 1H), 2.12 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ: 170.3, 169.0, 168.8, 67.4, 52.94, 52.89, 49.7, 48.0, 34.3, 29.7, 20.7; FTIR (ATR) 2956, 1730, 1436, 1368, 1345, 1214, 1151, 1061, 1030, 897, 844, 733, 702 cm−1; HRMS m/z (DART) Calcd for C10H16BrO6 (M + H)+ 311.0125. Found 311.0121.
Dimethyl 2-(2-Bromo-6-hydroxyhexyl)malonate (3k)26)3k (colorless oil); TLC (SiO2) Rf = 0.21 (n-hexane : ethyl acetate = 2 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.02–3.98 (m, 1H), 3.84 (dd, J = 5.7, 4.6 Hz, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 3.66 (t, J = 6.3 Hz, 2H), 2.49 (td, J = 7.5, 4.6, 3.4 Hz, 1H), 2.27 (td, J = 8.0, 6.9, 4.6 Hz, 1H), 1.94–1.86 (m, 2H), 1.69–1.49 (m, 5H); 13C{1H} NMR (125 MHz, CDCl3) δ: 169.3, 169.2, 62.5, 54.6, 52.82, 52.76, 50.2, 39.1, 37.8, 31.8, 23.7.
Diethyl 2-(2-Bromo-3-phenylpropyl)malonate (3l)27)3l (colorless oil); TLC (SiO2) Rf = 0.50 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.34–7.31 (m, 2H), 7.29–7.28 (m, 1H), 7.22–7.21 (m, 2H), 4.25–4.15 (m, 5H), 3.81 (dd, J = 6.3, 4.0 Hz, 1H), 3.25–3.17 (m, 2H), 2.52 (td, J = 6.9, 4.0, 2.9 Hz, 1H), 2.24 (td, J = 8.0, 4.0, 4.0 Hz, 1H), 1.28–1.23 (m, 6H); 13C{1H} NMR (125 MHz, CDCl3) δ: 168.9, 168.6, 137.7, 129.2, 128.5, 127.0, 61.7, 61.6, 54.1, 50.6, 45.8, 37.1, 14.0.
1,3-Diphenyl-2-(2-bromo-3-phenylpropyl)-1,3-propanedione (3m)3m (colorless oil); TLC (SiO2) Rf = 0.41 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 8.13 (d, J = 7.5 Hz, 2H), 7.91 (d, J = 7.5 Hz, 2H), 7.64 (t, J = 6.3 Hz, 1H), 7.53 (t, J = 7.5 Hz, 3H), 7.40 (t, J = 7.5 Hz, 2H), 7.32–7.29 (m, 2H), 7.26–7.25 (m, 1H), 7.23–7.21 (m, 2H), 5.83 (d, J = 10.9 Hz 1H), 4.41–4.37 (m, 1H), 3.31–3.19 (m, 2H), 2.88 (td, J = 10.9, 4.0, 2.3 Hz, 1H), 2.27–2.22 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ: 195.9, 195.0, 137.6, 136.1, 134.8, 134.0, 133.5, 129.2, 129.1, 128.9, 128.8, 128.4, 128.3, 127.0, 56.8, 54.9, 46.0, 37.9; FTIR (ATR) 3086, 3062, 3029, 2921, 2253, 1965, 1814, 1693, 1673, 1596, 1580, 1495, 1448, 1432, 1344, 1321, 1304, 1257, 1219, 1194, 1181, 1158, 1102, 1076, 1054, 1029, 990, 969, 943, 911, 887, 857, 822, 809, 786, 749, 734, 694, 652 cm−1; HRMS m/z (DART) Calcd for C24H22BrO2 (M + H)+ 421.0798. Found 421.0801.
1,3-Di-tert-butyl-2-(2-bromo-3-phenylpropyl)-1,3-propanedione (3n)3n (colorless oil); TLC (SiO2) Rf = 0.44 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.32 (t, J = 6.9 Hz, 2H), 7.273–7.271 (m, 1H), 7.20 (d, J = 6.9 Hz, 1H), 4.94 (dd, J = 8.6, 2.3 Hz, 1H), 3.95–3.90 (m, 1H), 3.23–3.11 (m, 2H), 2.52 (td, J = 5.7, 3.4, 2.3 Hz, 1H), 2.01 (td, J = 9.6, 5.2, 2.3 Hz, 1H), 1.28 (s, 9H), 1.11 (s, 9H); 13C{1H} NMR (125 MHz, CDCl3) δ: 211.7, 209.8, 137.5, 129.2, 128.4, 127.0, 56.4, 54.7, 45.9, 44.9, 44.4, 37.9, 28.3, 26.6; FTIR (ATR) 3778, 3064, 3030, 2966, 2910, 2872, 1715, 1692, 1604, 1497, 1479, 1456, 1421, 1395, 1367, 1338, 1286, 1227, 1193, 1156, 1098, 1057, 1006, 975, 942, 914, 864, 830, 801, 749, 699 cm−1; HRMS m/z (DART) Calcd for C20H30BrO2 (M + H)+ 381.1424. Found 382.1478.
Dimethyl 2-(2-Bromo-3-phenylpropyl)-2-methylmalonate (3o)3o (colorless oil); TLC (SiO2): Rf = 0.18 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.34–7.31 (t, J = 6.9 Hz, 2H), 7.28–7.26 (m, 1H), 7.22–7.20 (d, J = 7.8 Hz, 2H), 4.25–4.16 (m, 1H), 3.70 (s, 3H), 3.67 (s, 3H), 3.23–3.13 (m, 2H), 2.60 (d, J = 5.5 Hz, 2H), 1.46 (s, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ: 172.11, 172.06, 138.0, 129.3, 128.5, 127.0, 52.9, 52.8, 52.7, 50.7, 46.8, 43.5, 19.9; FTIR (ATR) 3029, 3000, 2953, 1733, 1603, 1497, 1455, 1435, 1381, 1315, 1257, 1201, 1167, 1116, 1030, 982, 928, 887, 850, 749, 701 cm−1; HRMS m/z (DART) Calcd for C15H20BrO4 (M + H)+ 343.0540. Found 343.0556.
Dimethyl 2-(2-Bromo-3-phenylpropyl)-2-propylmalonate (3p)3p (colorless oil); TLC (SiO2) Rf = 0.18 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.32 (t, J = 6.9 Hz, 2H), 7.29–7.27 (m, 1H), 7.20 (d, J = 7.5 Hz, 2H), 4.13–4.08 (m, 1H), 3.72 (s, 3H), 3.64 (s, 3H), 3.25–3.10 (m, 2H), 2.63–2.53 (m, 2H), 1.97 (td, J = 8.0, 4.6, 2.9 Hz, 1H), 1.79 (td, J = 9.7, 4.6, 4.0 Hz, 1H), 1.08–0.98 (m, 1H), 0.83 (t, J = 6.9 Hz, 3H), 0.78–0.68 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ: 171.6, 171.5, 138.0, 129.3, 128.5, 127.0, 56.6, 52.6, 52.5, 50.0, 47.0, 40.1, 34.3, 17.1, 14.2; FTIR (ATR) 3029, 2958, 2875, 1734, 1497, 1455, 1435, 1284, 1231, 1213, 1162, 1124, 1044, 937, 846, 750, 702 cm−1; HRMS m/z (DART) Calcd for C17H24BrO4 (M + H)+ 371.0853. Found 371.0857.
Dimethyl 2-(2-Bromo-3-phenylpropyl)-2-cyanoethylmalonate (3q)3q (colorless oil); TLC (SiO2) Rf = 0.46 (n-hexane : ethyl acetate = 4 : 1); 1H-NMR (500 MHz, CDCl3) δ: 7.38–7.7.34 (m, 2H), 7.30–7.29 (m, 1H), 7.21 (d, J = 7.5 Hz, 2H), 4.07–4.00 (m, 1H), 3.75 (s, 3H), 3.65 (s, 3H), 3.31–3.25 (m, 1H), 3.14–3.08 (m, 1H), 2.62–2.54 (m, 2H), 2.42–2.40 (m, 1H), 2.26–2.14 (m, 2H), 2.01–1.94 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ: 170.1, 170.0, 137.6, 129.3, 128.8, 127.3, 118.6, 55.6, 53.1, 53.0, 48.7, 47.0, 40.1, 28.2, 12.6; FTIR (ATR) 2956, 2254, 1732, 1437, 1378, 1206, 1093, 904, 726 cm−1; HRMS m/z (DART) Calcd for C17H21BrNO4 (M + H)+ 382.0649. Found 382.0661.
Dimethyl 2-((2,2,6,6-Tetramethylpiperidin-1-yl)oxy)malonate (5a)25)4a (white solid); TLC (SiO2) Rf = 0.40 (n-hexane : ethyl acetate = 10 : 1); 1H-NMR (500 MHz, CDCl3) δ: 4.97 (s, 1H), 3.79 (s, 6H), 1.51–1.25 (m, 6H), 1.20 (s, 6H), 1.06 (s, 6H); 13C{1H} NMR (125 MHz, CDCl3) δ: 167.7, 86.5, 60.4, 52.6, 40.1, 32.5, 20.1, 17.0; mp = 83.4 °C
The authors are thankful to the Takeda Science Foundation for financial supports.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.