Abstract
The crystal structures of two methoxyphenylbenzamide isomers are described, (Ph2Br) and (Ph3Br), with the general formula C14H12BrNO2. This structural study revealed the presence of N–H–O and C–H–O hydrogen bonds, Br–Br halogen bonds, C–H–π, and C–Br–π molecular contacts, showing in both compounds, a central C1–C7(O1)–N1(H1)–C8 amide segment, to be almost linear. The close proximity between the Br1 and O1 in Ph2Br showed that its interatomic distance was less than the sum of their VDW radii, generating an increase in the electrostatic potential in the O1 region, making possible the appearance of the so-called σ and π-holes on bromine. These specific conditions give rise to the formation of the Br–Br halogens bonds, which are united in a very interesting way, allowing the bond to extend by joining halogen atoms between different molecules forming an isosceles triangle with Br–Br distances equal to 3.5403(4) Å and 5.085 Å as its base. The presence of the carbonyl group in Ph2Br, an excellent acceptor of hydrogen and halogen bonds, led to competition between these bonds to organize crystal growth. The analysis of the compounds as pharmacophores showed that the bromine atom plays a key role in interactions with protein residues, reaching good ligand-protein interaction values comparable to the values presented by the parent inhibitor, Asciminib. In contact with the ALA356 residue, the bromine of Ph2Br participates with a higher contact geometry using the σ-hole, whereas the bromine of Ph3Br employs a more efficient contact geometry by taking advantage of its π-hole.
Introduction
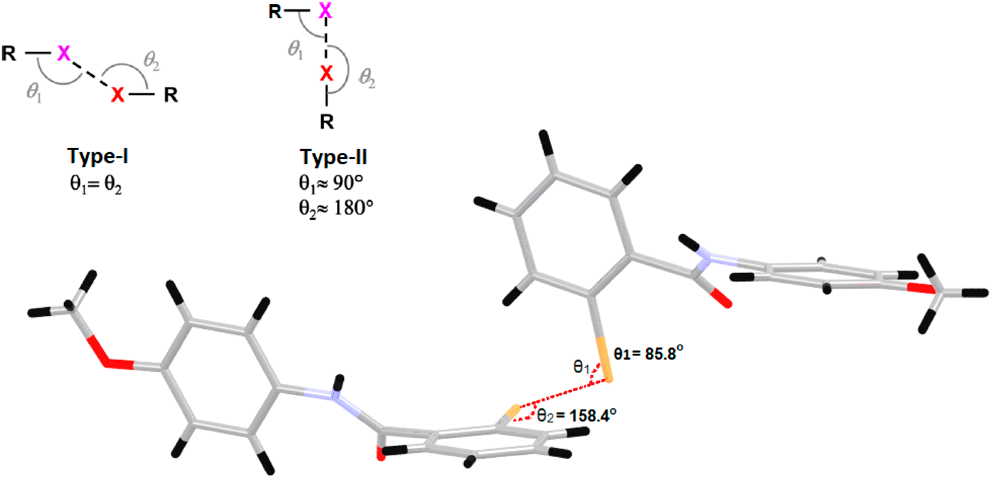
The study of crystal structures provides essential information about the geometry of the species that form the crystal, the symmetry relationships that characterize these species in the unit cell, and the different forms of bonding between the participating species. The literature shows that benzamide derivatives have antimicrobial,1) analgesic,2) anti-inflammatory,3) anticancer,4) and parasitic disease activities, among others.5,6) Halogen bonds also play an important role in crystal engineering,7) material chemistry,8) medicinal chemistry and biochemistry.9) Halogen bonding is considered one of the most important interactions in crystal growth and, in some cases, is shown to be competitive with hydrogen bonding.10) Hydrogen and halogen bondings play a special role in the arrangement of molecules in the crystal and are responsible for numerous physical, chemical, and biochemical processes.11,12) Generally, by using the X-ray diffraction techniques, numerous discoveries about the character and behavior of hydrogen and halogen bonding, the formation of molecular aggregates that allow the recognition of supramolecular patterns, synthons inherent to each structure have been associated with the study of crystal structures. The supramolecular study of a crystal structure suggests the hierarchy obeyed by the interactions present in it,13,14) including the weakest possible interactions, thus achieving a complete analysis of the crystal organization. The literature frequently shows the presence of halogen bonds from two different compounds, one of them acting as a halogen bond donor and the other as an acceptor; however, reports of halogen bonds emerging from a single molecule are much less frequently discussed.15,16) Medicinal chemistry has long tried to make small modifications in the structures of biologically active compounds to optimize their biological action. However, this objective is not always achieved since studies of structure–activity relationships have shown that chemically similar compounds can present significant differences in their biological actions.17) One of the most important parameters that affect biological activity depends on the flexibility of the molecules, a specific number of rotational bonds where it can adopt different geometries that favor the interactions that lead to higher energetic stability of the ligand. In recent years, there has been growing interest in the topic of non-covalent interactions involving σ or π holes. The relevance of these interactions is reflected in their high directionality and strength compared to hydrogen bonds. The Crystallography Research Group, GCRIS, has been deeply interested in the study of amido-halogenated compounds motivated by the crystalline and structural properties they may present.18) The goal of this work is to propose the synthesis of two amide organo-halogen compounds, which are the primary components of biomolecules, found in natural products or processed products such as pesticides or as pharmacophores that had the possibility of interacting with other molecules not only through hydrogen bonds but also through halogen bonds. The structures of these two amides are close to the structure of Asciminib,19) a pharmacophore used in patients with Chronic Myelogenous Leukemia.20) The presence of bromine atoms in the molecule’s structure opens the possibility of finding halogen bonds in its crystal growth and enabling halogen interactions with protein residues in active pockets. With C–X bond distance and polarizability being higher in bromine compared to fluorine or chlorine, it can be advantageous in forming relatively strong halogen bonds with different Lewis bases in an active site.21) Halogen bonds of the type Br–Br or weaker C–Br–π contacts that are fundamental to the design of its crystal arrangement are proposed. Contacts like C–X–X–C, where X is the halogen atom, according to the angles θ1 and θ2, are classified into type I or II, where a type I contact is believed to be a consequence of crystal packing effects, while type II is a product of polarization effects of the halogen atoms in the crystal.22,23) Following the definition of halogen bonding (IUPAC Recommendations 2013), it can be taken as a net attractive interaction between an electrophilic region associated with the halogen atom in one molecular entity with another nucleophilic region in another molecular entity, or it can be in the same molecular entity in case of intramolecular interactions.24) Halogen interactions can form an electropositive region called σ-hole, where there is a deficiency in the electron density due to the anisotropy of the system involved. The magnitude of the σ-hole is directly proportional to the polarizability of the halogen atom involved, noting that the bromine atom occupies an intermediate value between the polarizabilities of the atoms in group VII.25) Real-space imaging of the anisotropic σ-hole charge was recently reported.26) This work aims to design two aryl halide isomers containing bromine atoms in the ortho and meta positions of their rings, defining the most important interactions that control their crystal growth together with the role played by the halogen bond in defining the properties of each of the isomers.
Results and Discussion
The crystal structures of these organo-halogen compounds are determined and analyzed in terms of N–H–O, C–H–O, C–H–π, Br–Br and C–Br–π hydrogen or halogen bonding and halogen contacts.12,27) A good strategy for these applications in supramolecular studies can be followed by following the hierarchy of interactions present in its crystal structure,13,14) finding the presence of different synthons and weaker π–π interactions that help to define more appropriately their molecular growth. The GCRIS group of the Universidad del Valle has oriented its efforts on obtaining single-crystal compounds suitable for X-ray diffraction (XRD), focusing on their structural, supramolecular, spectroscopic, and theoretical elucidation. It was therefore proposed, in the present research work, the synthesis of two isomers: 2-bromo-N-(4-methoxyphenyl)benzamide (Ph2Br) and 3-bromo-N-(4-methoxyphenyl)benzamide (Ph3Br) (Fig. 1), very close in their physical and chemical properties, with identical structural flexibility and that can be compared to the structure of the drug Asciminib.19) With the aim of analyzing their properties, similarities, and dissimilarities in their behavior, theoretical studies were undertaken that contribute to revealing the advantages of one of these compounds for a possible biological application. Among their similarities, the compounds have a central C1–C7(O1)–N1(H1)–C8 amide segment that is almost planar with RMS deviation of 0.0151 and 0.0068 Å for the ortho and meta systems, respectively, forming dihedral angles with the aromatic rings of 45.16(4)° and 34.97(4)° in Ph2Br and 31.09(10)° and 37.90(10)° in Ph3Br.
Preliminary results show that although the two titled compounds possess similar physical and chemical properties, they differ considerably in the unit cell parameters and in the crystalline growth due to different interactions that each molecule presents in the growth process.
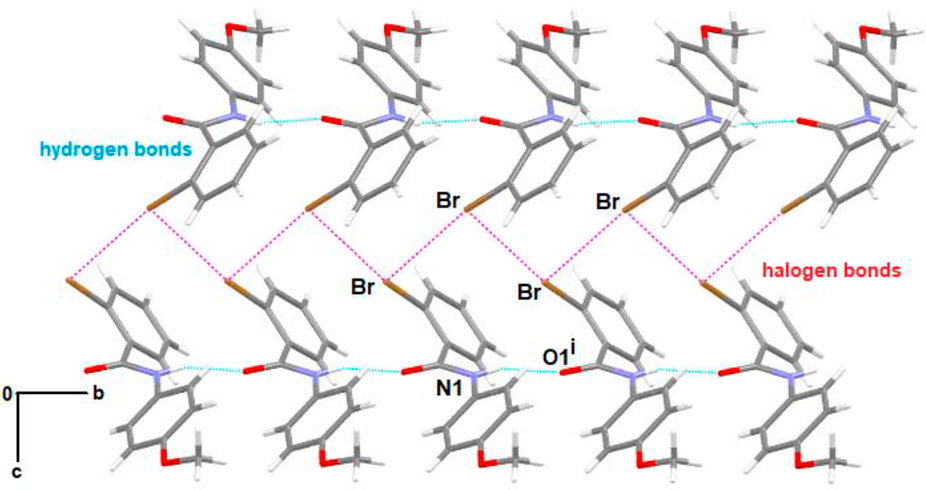
Supramolecular AnalysisThe structural analysis of the compounds was carried out using the Parst28) and Platon29) programs and was complemented with three-dimensional modeling using Mercury30) and CrystalExplorer.31) Hydrogen bonding, an attractive and stabilizing interaction between a hydrogen donor D–H group and a more electronegative acceptor atom, provides directionality and stabilization to the crystal arrangement, which together with halogen bonding are non-covalent interactions of vital importance for the arrangement of the molecules during the crystallization process. Table 1 shows the distances and angles involved in the interactions that control crystal growth in Ph2Br and Ph3Br compounds Fig. 2 partially shows the crystal growth of Ph2Br highlighting the N–H–O and Br–Br interactions following the [010] direction. The supramolecular growth of Ph2Br is strongly dependent on N–H–O interactions inherent to this type of amide compound. Along b, for example, N1–H1–O1i interactions, where the N1–H1 group (x, y, z) acts as a hydrogen donor concerning the O1i atom in the molecule at (x, y + 1, z), strongly affect the crystal growth. These interactions are supported by halogen bonds (C–Br–Br) that contribute to the crystal growth along [001] (Fig. 2). These halogen bonds are bound in a very interesting way, allowing the bond to extend by joining halogen atoms between different molecules along [010], forming an isosceles triangle with Br–Br distances equal to 3.5403(4) Å on their adjacent sides and 5.085 Å at its base. This arrangement ensures a special structure that enables higher stability and establishes its own seal on the growth process. The role of Br atoms does not conclude here, as they actively participate by interacting with molecular regions having π clouds, forming C–Br–Cg1iii bonds. Additionally, weak C–H–π interactions contribute positively to the crystallization process (Fig. 3).
Table 1. Most Relevant Interactions in Supramolecular Growth for
Ph2Br and
Ph3Br Compounds
| System | D–H–A | D–H or D–Br | H–A or Br–A | D–A | D–H–A |
|---|
| Ph2Br | N1–H1–O1i | 0.86 | 2.08 | 2.8788(14) | 153.3 |
| C12–H12–Cg2ii | 0.93 | 2.902 | 3.646 | 137.88 |
| C2–Br1–Cg1iii | 1.8896(11) | 3.6193(7) | 4.212 | 94.48 |
| Br–Briv | — | 3.7085(6) | 3.5403(4) | — |
| Ph3Br | N1–H1–O1v | 0.86 | 2.32 | 3.157(3) | 165.7 |
| C5–H5–Cg3vii | 0.93 | 2.960 | 3.674 | 134.72 |
| C14–H141–O2vi | 0.960 | 2.67 | 3.524(4) | 147.9 |
Distances are given in Å. Symmetry codes: (i) x, y + 1, z; (ii) 1/2−x, 1/2 + y, 1/2−z; (iii) x, y−1, z; (iv) 3/2−x, −1/2 + y, 1/2−z; (v) x, −y + 3/2, + z−1/2; (vi) −x + 1, −y + 1, −z + 2.
The supramolecular analysis of the two isomers showed that Ph2Br was the isomer that clearly presented the halogen bond, where Bri acts as an acceptor of a charge density coming from Brii. Theoretical calculations support this reasoning, and several authors have cataloged the halogen interaction as a true bond, which provides directionality and geometry in crystal growth.32) Theoretical and experimental studies have shown that halogen bonding is much more directional than hydrogen bonding since the σ-hole is located just at the elongation of the covalent bond to which the halogen is attached.33) The halogen bond geometry is classified according to the value of the angles θ1 and θ2, as presented in Fig. 4. The Ph2Br system was classified as type II since it fulfills the condition θ1 ≈ 90° and θ2 ≈ 180°.
Two organo-halogen compounds isomorphous with Ph2Br were deposited in the CCDC database, with the numbers 1963339 and 799713. These two crystalline systems with very close cell parameters and equal space group, present very similar intermolecular interactions; however, these two compounds do not present halogen intermolecular bonds, a parameter that is the seal that characterizes Ph2Br.
For the Ph3Br, no halogen bonds are observed, emphasizing the N–H–O and C–H–O interactions strongly contribute to the crystal growth. The supramolecular growth of Ph3Br is determined by a very interesting set of hydrogen bonds. Along b particularly, N1–H1–O1v interactions are observed, where the N1–H1 (x, y, z) group acts as a hydrogen bond donor for the O1v atom at (x, −y + 3/2, + z−1/2), resulting in the formation of chains of molecules that constitute the backbone of the system. This interaction is supported by the formation of constituted dimers, which are joined in a very particular way to connect the ribbons by hydrogen bonds C14–H14–O2vi, where the C14–H14 group (x, y, z) acts as a hydrogen bond donor for the O2vi atom at (−x + 1, −y + 1, −z + 2) forming fused R22(6) ring motifs (Fig. 5). Finally, C–H–π interactions are observed between the bromine–benzene rings at a distance of 3.638 Å, strengthening the scaffold of the crystal structure.
It is important to emphasize the relevance of the interactions in both systems since it is fascinating to observe that a simple change in the position of a bromine atom in the ring can result in totally different spatial arrangements and probably in very different properties.
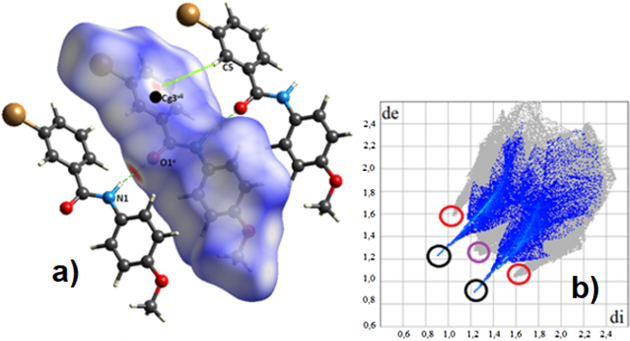
Hirshfeld Surface AnalysisThe fidelity of the intermolecular interactions studied in the previous section was visualized using Hirshfeld surface (HS) analysis. The study of the HS was carried out using the CrystalExplorer program31) to visualize and analyze the interactions present in the crystal. In the analysis of the intermolecular contacts, the surface called dnorm was used, highlighting the most relevant interactions in the compounds. This surface contains the parameters di (distance from the surface to the nearest nucleus inside it), de (distance from the surface to the nearest nucleus outside it), and the van der Waals radii (VDWr) of the atoms. The surface shows the closest contacts in red, the ones in white are those with a distance equivalent to the VDWr, and the ones in blue offer the most distant contacts. The combination of the de and di parameters form the so-called two dimensional (2D) fingerprint, which is unique and representative of each molecule. These plots are used to quantify the contribution of each interaction present in the crystal.
The analysis of these surfaces is of utmost importance as it helps to check in detail the key players in crystal growth. In the Ph2Br compound, together with the N1–H1–O1i interaction, which stands out as one of the closest and strongest contacts on the surface along b, Br–Briv interactions are observed, showing the directionality of the halogen bonding. Other interactions, C2–Br1–Cg1iii and C12–H12–Cgii, despite being weak interactions, bring higher stability to the crystalline arrangement (Fig. 6a). We obtain the so-called fingerprint by performing a de vs. di mapping on the surface (Fig. 6b). Hirshfeld surface analysis shows that O–H/H–O interactions appear as symmetric peaks that are highlighted in black circles, compromising 16.9% of the surface. In turn, the C–H/H–C interactions appear as clamps highlighted in red circles, making up 29.0% of the surface area. The Br–Br interaction highlighted in yellow compromises 2.8% of the surface, while the H–H interaction highlighted with a violet circle and made up 34.6% of the surface. The dnorm surface results match the fingerprint plot of the system.
As observed in Fig. 7a, the N1–H1–O1v bond acts as a backbone providing support and stability in its molecular growth along b. On the other hand, the formation of molecular dimers due to the C14–H14–O2vi along b is also observed. Finally, weak C5–H5–Cg3vii interactions are present, which help to stabilize the stacking of the bromobenzene rings. Hirshfeld surface analysis of Ph3Br shows that O–H/H–O interactions, highlighted in black circles, compromise 15.0% of the surface (Fig. 7b). Additionally, C–H/H–C interactions, which appear as side clamps, highlighted in red circles, make up 32.5% of the surface area. Finally, H–H interactions, highlighted in a violet circle, make up 30.9% of the surface. The dnorm surface results match the fingerprint plot of the system.
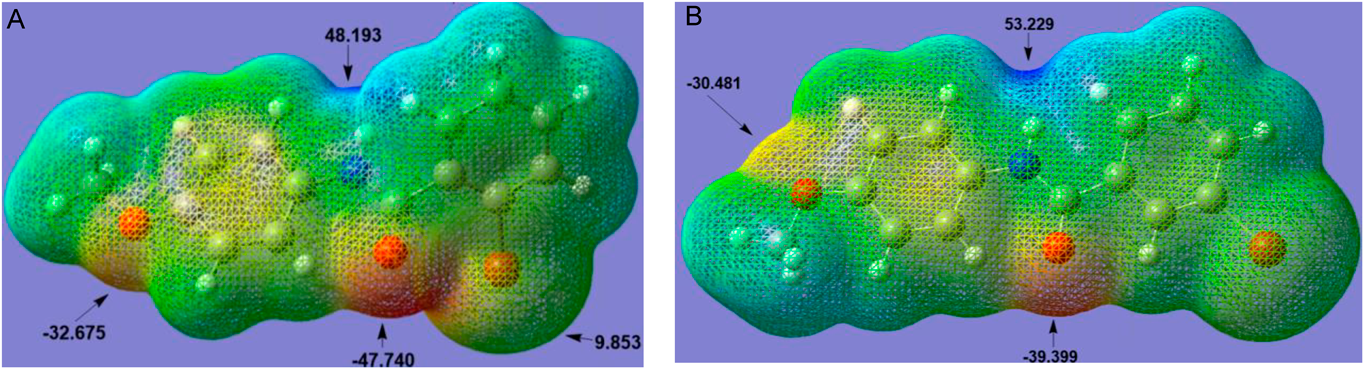
Molecular Electrostatic Potential (MEP)The MEP is considered as the potential that a unit of positive charge would experience at some point around the molecule due to its electronic distribution. To understand the nature and origin of the driving forces of crystal growth, a quantitative analysis of the electrostatic potential surface for Ph2Br and Ph3Br molecules was performed using Multiwfn software, a multifunctional program for wave function analysis.34) Regions with high negative potentials have a high probability of undergoing electrophilic attacks or experiencing protonation, whereas high positive potentials indicate regions with low electron density prone to nucleophilic attack. The MEPs of Ph2Br and Ph3Br compounds are shown in Fig. 8, categorizing the leading role of the amide group in the crystal growth of both isomers.
Both the oxygen atom of the carbonyl group (O1) and the hydrogen atom directly bonded to the nitrogen atom (N1) have the most relevant electrostatic potential values (−47.7 and 48.2 kcal/mol) for Ph2Br and (−39.4 and 53.2 kcal/mol) for Ph3Br. These, being the most representative values, constitute the directing centers of the crystalline growth in both isomers. The study also shows, for Ph2Br, the presence of an electro-deficient region, close to the bromine atom, of approximately 9.8 kcal/mol. The distance between O1 and Br1 in Ph2Br shows a value of 3.126 Å, which is smaller than the sum of the VDWr (3.37 Å).35) The high potential generated by the carbonyl group (−47.7 kcal/mol) acts on the bromine atom, which is highly polarizable, causing an anisotropic perturbation of its electron density, creating electro-deficient regions that would lead to the appearance of σ and π holes, making them fundamental for the interactions they will undertake with bromine atoms of neighboring molecules (Fig. 2). Figure 8 also shows the attractive character of the oxygen atom of the methoxy group in both isomers −32.7 kcal/mol (Ph2Br) and −30.5 kcal/mol (Ph3Br) respectively. The oxygen atom of the methoxy group plays an important role in both isomers as it contributes energetically to a possible electrostatic stabilization. It is interesting to analyze these results through possible interactions that the compounds may have as inhibitors in an active protein pocket.
Energy Structure AnalysisThe CrystalExplorer program makes it easier to obtain the three-dimensional visualization of the interactions in a molecular crystal by manipulating its Hirshfeld surfaces. It can also generate the fingerprint plots (2D) of these interactions.31) This program analyses the energies of molecular interactions in a crystalline compound and is based on the “PIXEL” model,36) which analyzes the molecules in the crystal as a whole, as opposed to analysis through intermolecular atom-atom contacts. The energy values can be described as the sum of electrostatic (Eele), polarization (Epol), dispersion (Edis), and repulsion (Erep) energies.37)
The interaction energies of the selected molecular pairs for Ph2Br in the first coordination sphere of radius 3.8 Å are shown in Table 2 and are visualized in Fig. 9.
Table 2. Molecular Pairs and Interaction Energies (kJ/mol) Obtained from the Solid-State Energy Analysis for the
Ph2BrThese results show that the most energetic interaction (Etot=−63.5 kJ/mol) with a distance between the centroids of the rings, R = 5.08 Å, has a slightly higher dispersion energy component than the electrostatic component. This increase in the dispersion component is caused possibly by the C2–Br1–Cg1iii contacts, in which the σ-hole interacts with the π-cloud of the aromatic ring, along with the C–H–π interactions present in the crystal lattice. On the other hand, the electrostatic energy component would be due to N1–H1–O1i interactions, which serve as a counterpart in the equilibrium of forces for crystal growth along b. The stabilizing power of the latter interactions contributes to 32.9% of the total energy. The second most important energetic interaction (Etot=−43.6 kcal/mol) occurs with the symmetrically related molecule (−x, −y, −z) at R = 6.56 Å. This interaction shows that the dispersion energies are twice the electrostatic character energies (Edis=−42.2 kcal/mol and Eele=−22.6 kcal/mol). This behavior is due to the formation of C–H–π interactions in the aromatic rings, which prevails over the C–H–N interactions.
A third meaningful interaction with a total energy of −28.5 kcal/mol at R = 5.43 Å of the original molecule shows an energetic component where dispersion forces predominate. Finally, interactions with molecules of symmetry (−x + 1/2, y + 1/2, −z + 1/2) and a distance R = 8.69 Å, possibly corresponding to C12–H12–Cg2 interactions, contribute 10.6% of the total energy. The results of the interaction energies for Ph3Br are shown in the supplementary section.
Molecular DockingProtein-ligand docking has recently emerged as an effective tool for predicting ligand orientation when bound to a protein receptor. The potential of protein-ligand interactions, whether coulombic, van der Waals, or hydrogen bonding, is estimated by values showing the affinity of the ligand in the active region and is presented by a docking score.38) The partial structural similarity of Ph2Br and Ph3Br compounds to Asciminib, a newly U. S. FOOD & DRUG ADMINISTRATION (FDA)-approved drug for patients in the chronic phase of Philadelphia chromosome-positive chronic myeloid leukemia (Ph+CML) (CP),19) prompted us to evaluate the behavior of our compounds as inhibitors of ABL1 kinase-type proteins. Molecular docking studies were performed using AutoDockVina software,39) on the protein (PDB ID 5MO4) that was taken as a reference and integrated with the Asciminib molecule in co-crystallized form.40) As shown in Fig. 10, the inhibitor Asciminib (shown in wires) and Ph2Br and Ph3Br (indicated in blue and green) present similar features in their structures, the amide and ether bonds. In the drug Asciminib, the most important interaction with the active site corresponds to a bond between the terminal halogen of the ether group and the amino acid LEU448, being observed additionally two other interactions: a hydrogen bond between the pyrazole ring (N–H) and the amino acid (C=O) GLU481 and an aromatic interaction between the pyridine ring (Asciminib) and the amino acid TYR454. Due to the structural similarity with Asciminib, the proposed candidates adopt a favorable active site geometry. However, it is evident that, in Asciminib, the positive interactive contribution is achieved with the halogen atom.
It can be observed that Ph2Br and Ph3Br, retain the interaction of the aromatic ring with TYR454. A new contact with the ALA356 residue is revealed, where the bromine of Ph2Br engages in a higher contact geometry using an interaction parallel to the molecular axis, the σ-hole. In contrast, the bromine of Ph3Br adopts a binding geometry perpendicular to the molecular axis, the π-hole. The values obtained for the affinity energies were −7.5 and −7.7 kcal mol−1 for Ph2Br, and Ph3Br, respectively, versus −10.4 kcal mol−1 for Asciminib. Additional simulation studies were carried out on the in vitro/in vivo behavior of some halogenated benzamides as cyclooxygenase-1 (COX-1) inhibitors.41) For this purpose, the simulation of the behavior of our halogenated benzamides in the crystalline structure of the macromolecule (1PGE) (prostaglandin H2 synthase-1) with the ligand (iodosuprofen) in the active site, was considered.42) The affinity values found for the studied molecules were −7.7 kcal mol−1 for Ph2Br and −7.4 kcal mol−1 for Ph3Br, while iodosuprofen showed energy of −8.6 kcal mol−1 (Fig. 11). These relatively close energy values of the two pharmacophores show good adaptability in the active protein pocket and reveal the positive role played by the halogen in this inhibition process.
Conclusion
The position of the halogen atom did not provide any significant differences in bond distances in the isomers. However, the two organo-halogen amido structures showed the formation of dihedral angles between the central amide segment C1–C7(O1)–N1(H1)–C8 and the aromatic rings of the compounds of 45.16(4)° and 34.97(4)° in Ph2Br and 31.09(10)° and 37.90(10)° in Ph3Br.
The presence of the carbonyl group, an excellent acceptor of hydrogen and halogen bonds, promotes the formation of Br–Br bonds in Ph2Br forming isosceles triangles along [010], which are a critical part of its crystal growth.
The position of the bromine atom in the Ph2Br ring radically influenced the formation of Br–Br halogen bonds, thereby providing a defined directionality in its crystallization process.
The proximity of the bromine atom to the carbonyl group in Ph2Br is evidenced by an interatomic distance of O1–Br1 less than the sum of their respective van der Waals radii. This generates an increase in the electrostatic potential in the region of the O1 atom, producing in turn, an anisotropic distribution of the electron density in a specific region of the bromine atom, as shown by a higher electron deficiency than its surroundings, thus leading to the appearance of the so-called σ and π holes. All of these give rise to the emergence of the halogen Br–Br bonds.
The energy calculations generated by interactions between pairs of molecules showed that the Ph2Br isomer exhibited a 39% higher repulsion energy than Ph3Br, due to the observed proximity between the O1 and Br1 atoms and forming halogen bonds that force the molecules even closer together.
The analysis of the compounds as pharmacophores showed that the bromine atom plays a very important role in the interactions with protein residues, reaching good protein-ligand interaction values. In contact with the ALA356 residue, the bromine of Ph2Br participates with a higher contact geometry using the σ-hole, whereas the bromine of Ph3Br takes advantage of the π-hole.
Experimental
A solution of 4-methoxyaniline (1.00 g, 8.12 mmol) in chloroform added 2-bromobenzoyl or 3-bromobenzoyl chlorides in excess in a 2 : 1 ratio at 61 °C for four h. The reactions were magnetically stirred and monitored by TLC. After completion of the reactions, the remaining solids were filtered and crystallized by slow evaporation at room temperature in THF to obtain colorless crystals.
2-Bromo-N-(4-methoxyphenyl)benzamide1H-NMR (400 MHz, Chloroform-d) δ = ppm: 7.63 (ddd, J = 7.8, 5.9, 1.5 Hz, 1H), 7.61 (br, 1H), 7.54 (d, J = 9.0 Hz, 2H, MeO-Ar), 7.40 (td, J = 7.5, 1.2 Hz, 1H), 7.31 (td, J = 7.7, 1.8 Hz, 1H), 6.91 (d, J = 9.0 Hz, 2H, MeO-Ar), 3.82 (s, 3H, −CH3). 13C-NMR (100 MHz, CDCl3) δ = ppm: 165.51, 157.02, 138.03, 133.63, 131.69, 130.73, 130.00, 127.87, 122.10, 119.43, 114.45, 77.48, 76.84, 55.68. IR (FT-IR) cm−1: 519, 563, 640, 687, 749, 820, 903, 953, 1024, 1010, 1176, 1235, 1311, 1411, 1458, 1520, 1593, 1653, 2895, 2951, 3283.
3-Bromo-N-(4-methoxyphenyl)benzamide1H-NMR (400 MHz, Chloroform-d) δ = ppm: 7.67 (br, 1H), 7.62 (d, J = 7.7 Hz, 2H), 7.54 (d, J = 8.9 Hz, 2H, MeO-Ar), 7.38 (td, J = 7.5, 1.2 Hz, 1H), 7.30 (td, J = 7.7, 1.7 Hz, 1H), 6.90 (d, J = 8.9 Hz, 2H, MeO-Ar), 3.81 (s, 3H, −CH3). 13C-NMR (100 MHz, CDCl3) δ = ppm: 165.54, 156.98, 138.02, 133.59, 131.65, 130.75, 129.93, 127.83, 122.10, 119.43, 114.41, 77.48, 77.16, 76.84, 55.67. IR (FT-IR) cm−1: 519, 563, 640, 687, 749, 820, 903, 953, 1024, 1010, 1176, 1235, 1311, 1411, 1458, 1520, 1593, 1653, 2895, 2951, 3283.
X-Ray Crystal Structure AnalysesAPEX2 and SAINT,43) and SHELXL44) programs were used to determine and refine the structures of Ph2Br and Ph3Br. The ORTEP-3 for Windows45) was used to draw the thermal displacement ellipsoid plots at the 50% probability level.
Crystal DataFor Ph2Br C14H12BrNO2 (M = 306.16 g/mol): monoclinic, space group P 21/n (No. 14), a = 13.0590(7) Å, b = 5.0849(4) Å, c = 18.7393(17) Å, β = 98.994(7)°, V = 1229.06(16) Å3, Z = 4, T = 293 K, μ(MoKα) = 3.337 mm−1, Dcalc = 1.655 Mg/m3, 23611 reflections measured (2.903° ≤ θ ≤ 33.063°), 4625 unique, Rint = 0.028. The final R1 was 0.0241 (I > 2_(I)) and wR2 was 0.0599 (all data), S = 1.081. Correctness of the model was confirmed by low residual peaks (0.577) and holes (−0.831) e.Å−3.
For Ph3Br = C14H12BrNO2 (M = 306.16 g/mol): monoclinic, space group P 21/n (No. 14), a = 28.2433(10) Å, b = 5.28330(10) Å, c = 8.3371(2) Å, β = 91.4680(10)°, V = 1243.64(6) Å3, Z = 4, T = 293 K, μ(MoKα) = 3.298 mm−1, Dcalc = 1.635 Mg/m3, 2613 reflections measured (3.924° ≤ θ ≤ 25.709°), 2336 unique, Rint = 0.053. The final R1 was 0.0375 (I > 2_(I)) and wR2 was 0.0946 (all data), S = 1.008. Correctness of the model was confirmed by low residual peaks (0.468) and holes (−0.525) e.Å−3.
RefinementAll H atoms were found in Fourier-difference maps and placed in geometrically idealized positions using a riding model with C–H = 0.93 Å (ring), 0.96 Å (methyl), and N–H = 0.86 Å. All H atoms were refined with isotropic displacement parameters set to 1.2 times the Ueq(C, N) for the aromatic and amine group and 1.5 times the Ueq(C) for the methyl groups.46) All bond lengths and bond angles are within normal ranges.47) Supplementary Table S1 gives selected crystallographic parameters, data collection, and structure refinement details.
Computational MethodologyAll computational procedures were performed using Gaussian 09,48) and Gaussian view 6 programs.49) The optimized structure and additional calculations of both compounds were performed with B3LYP/6-31G(d,p) level of theory and DFT method, in the gas phase.
Acknowledgments
RMF thanks the Universidad del Valle, Colombia, for granting a sabbatical year and KAD thanks Universidad del Valle for supporting his doctoral studies.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
References
- 1) Straniero V., Suigo L., Casiraghi A., Sebastián-Pérez V., Hrast M., Zanotto C., Zdovc I., Morghen C. G., Radaelli A., Valoti E., Antibiotics., 9, 160 (2020).
- 2) Sakr A., Kothayer H., Ibrahim S. M., Baraka M. M., Rezq S., Bioorg. Chem., 84, 76–86 (2019).
- 3) Donnier-Marechal M., Carato P., Larchanché P.-E., Ravez R., Boulahjar R., Barczyk A., Oxombre B., Vermersch P., Melnyk P., Eur. J. Med. Chem., 138, 964–978 (2017).
- 4) Chen T., Jiang H., Zhou J., Li Z., Huang W., Luo Y., Zhao Y., Med. Chem., 16, 555–562 (2020).
- 5) Asif M., Mod. Chem. Appl., 4, 194–196 (2016).
- 6) Pastrana M., Surmay V., Galeano E., Robledo S., J. Braz. Chem. Soc., 30, 116–123 (2019).
- 7) Mukherjee A., Tothadi S., Desiraju G. R., Acc. Chem. Res., 47, 2514–2524 (2014).
- 8) Priimagi A., Cavallo G., Metrangolo P., Resnati G., Acc. Chem. Res., 46, 2686–2695 (2013).
- 9) Scholfield M. R., Van der Zanden C. M., Carter M., Ho P. S., Protein Sci., 22, 139–152 (2013).
- 10) Metrangolo P., Neukirch H., Pilati T., Resnati G., Acc. Chem. Res., 38, 386–395 (2005).
- 11) Grabowski S. J., Phys. Chem. Chem. Phys., 19, 29742–29759 (2017).
- 12) Samai S., Biradha K., CrystEngComm., 11, 482–492 (2009).
- 13) Etter M. C., Acc. Chem. Res., 23, 120–126 (1990).
- 14) Etter M. C., J. Phys. Chem., 95, 4601–4610 (1991).
- 15) Hettstedt C., Mayer P., Karaghiosoff C., New J. Chem., 39, 8522–8533 (2015).
- 16) Zierkiewicz W., Michalczyk M., Scheiner S., Molecules, 26, 1740 (2021).
- 17) Kubinyi H., J. Braz. Chem. Soc., 13, 717–726 (2002).
- 18) Moreno-Fuquen R., Hurtado-Angulo M., Arango-Daraviña K., Bain G., Kennedy A. R., Acta Crystallogr. Sect. E Crystallogr. Commun., 76, 1762–1767 (2020).
- 19) Hughes T. P., Mauro M. J., Cortes J. E., et al., N. Engl. J. Med., 381, 2315–2326 (2019).
- 20) Miura M., Biol. Pharm. Bull., 38, 645–654 (2015).
- 21) Wilcken R., Zimmermann M. O., Lange A., Joerger A. C., Boeckler F. M., J. Med. Chem., 56, 1363–1388 (2013).
- 22) Pedireddi V. R., Reddy D. S., Goud B. S., Craig D. C., Rae A. D., Desiraju G. R., J. Chem. Soc., Perkin Trans. 2, 2, 2353–2360 (1994).
- 23) Desiraju G. R., Parthasarathy R., J. Am. Chem. Soc., 111, 8725–8726 (1989).
- 24) Desiraju G. R., Shing Ho P., Kloo L., Legon A. C., Marquardt R., Metrangolo P., Politzer P., Resnati G., Rissanen K., Pure Appl. Chem., 85, 1711–1713 (2013).
- 25) Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev., 116, 2478–2601 (2016).
- 26) Mallada B., Gallardo A., Lamanec M., De la Torre B., Spirko V., Hobza P., Jelinek P., Science, 374, 863–867 (2021).
- 27) Matsumoto A., Tanaka T., Tsubouchi T., Tashiro K., Saragai S., Nakamoto S., J. Am. Chem. Soc., 124, 8891–8902 (2002).
- 28) Nardelli M., J. Appl. Cryst., 28, 659 (1995).
- 29) Spek A. L., Acta Crystallogr. D Biol. Crystallogr., 65, 148–155 (2009).
- 30) Macrae C. F., Edgington P. R., McCabe P., Pidcock E., Shields G. P., Taylor R., Towler M., van de Streek J., J. Appl. Cryst., 39, 453–457 (2006).
- 31) Spackman P. R., Turner M. J., McKinnon J. J., Wolff S. K., Grimwood D. J., Jayatilaka D., Spackman M. A., J. Appl. Cryst., 54, 1–6 (2021).
- 32) Politzer P., Murray J. S., Clark T., Phys. Chem. Chem. Phys., 12, 7748–7757 (2010).
- 33) Kolár M., Hostas J., Hobza P., Phys. Chem. Chem. Phys., 16, 9987–9996 (2014).
- 34) Lu T., Chen F., J. Comput. Chem., 33, 580–592 (2011).
- 35) Bondi A., J. Phys. Chem., 68, 441–451 (1964).
- 36) Gavezzotti A., Zeitschrift fur Krist., 220, 499–510 (2005).
- 37) Mackenzie C. F., Spackman P. R., Jayatilaka D., Spackman M. A., IUCrJ., 4, 575–587 (2017).
- 38) Pagadala N. S., Syed K., Tuszynski J., Biophys. Rev., 9, 91–102 (2017).
- 39) Trott O., Olson A. J., J. Comput. Chem., 31, 455–461 (2010).
- 40) Wylie A. A., Schoepfer J., Jahnke W., et al., Nature (London), 543, 733–737 (2017).
- 41) Kakuta H., Zheng X., Oda H., Harada S., Sugimoto Y., Sasaki K., Tai A., J. Med. Chem., 51, 2400–2411 (2008).
- 42) Loll P. J., Picot D., Ekabo O., Garavito R. M., Biochemistry, 35, 7330–7340 (1996).
- 43) Bruker (2014). XPREP (Version 2014/2) and SADABS (Version 2014/4). Bruker AXS Inc., Madison, Wisconsin, U.S.A.
- 44) Sheldrick G. M., Acta Crystallogr. C, 71, 3–8 (2015).
- 45) Farrugia L. J., J. Appl. Cryst., 45, 849–854 (2012).
- 46) Lübben J., Volkmann C., Grabowsky S., Edwards A., Morgenroth W., Fabbiani F. P. A., Sheldrick G. M., Dittrich B., Acta Crystallogr. A, 70, 309–316 (2014).
- 47) Allen F. H., Kennard O., Watson D. G., Brammer L., Guy Orpen A., Taylor R., J. Chem. Soc., Perkin Trans. 2, S1–S19 (1987). https://pubs.rsc.org/en/content/articlelanding/1987/p2/p298700000s1
- 48) Gaussian 09, Revision E.01, Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J. Gaussian, Inc., Wallingford CT, 2009. n, Inc., Wallingford CT, 2016.
- 49) GaussView, Version 6, Dennington, Roy; Keith, Todd A.; Millam, John M. Semichem Inc., Shawnee Mission, KS, 2016. Gaussian View 09. 2009.