Abstract
Diverse naturally occurring events relevant to proteins, including processing and post-translational modification, provide significant clues enabling advances in peptide/protein chemistry. Our motivation to synthesize large proteins prompted us to seek scientific bases utilized in synthetic experiments on the intein-mediated protein processing system. This account describes peptide/protein thioester-producing protocols whose design is based on acyl transfer reactions observed in the intein system, and the development of the stimulus-responsive amide cleavage and its application to the modulation of peptide function. Finally, several findings derived from nature-inspired research efforts, including peptide mimetic synthesis and S-protected cysteine chemistry, are described.
1. Introduction
The natural protein-editing system, the intein system,1–5) has provided much insight for development of various chemical possibilities for peptide/protein chemistry. Three acyl transfer reactions which operate sequentially in the intein system accomplish the self-editing of intein-separated protein sequences and release the intervening intein region (Fig. 1). Chemistry-guided mimicking of the intein system allowed us to enrich this chemical toolbox, thus advancing peptide/protein chemistry. Newly developed tools include chemical devices for thioester synthesis, protein thioester-producing systems, stimulus-responsive amide-cleaving systems and various chemical means derived from the findings in this research. This account summarizes the new chemical tools and their practical use in peptide/protein chemistry.
2. The Intein System
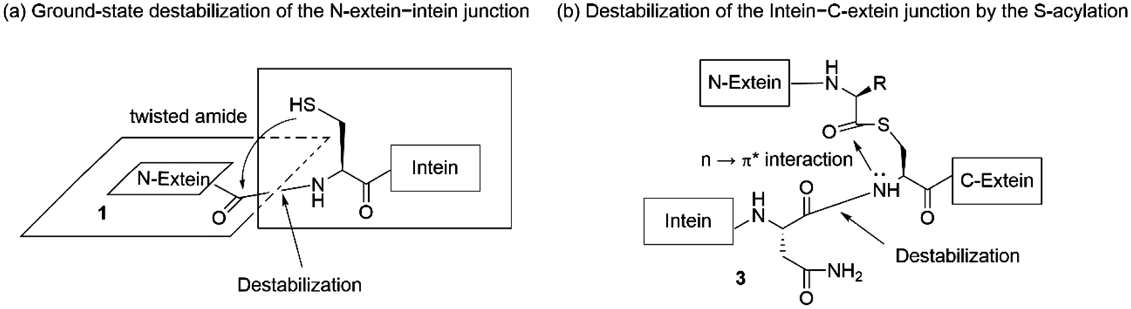
Sequential N–S, S–S and S–N acyl transfer of an intein-containing protein 1 completes the protein processing with the intein sequence 4 excised from the parent protein 1 to yield a mature protein 6. Here, the N-extein and C-extein sequence which has been split by the intein develops into a mature protein 6 consisting of N- and C-exteins. This takes place by the self-splicing action of the intein via the intermediates 2, 3 and 54,5) (Fig. 1). Among the three acyl/peptidyl transfer steps involved, mimicking the first (1. N–S acyl transfer) and the third (3-a. amide cleavage and 3-b. S–N acyl transfer) steps led to the development of chemical protocols which allow protein chemical synthesis, protein thioester synthesis and stimulus-triggered peptide manipulation. Thermodynamically unfavored conversion of amides 1 to thioesters 2 occurs in the first acyl transfer step, in which the amide bond suffers ground-state destabilization and becomes susceptible to the nucleophilic attack by the sulfenyl group of the neighboring cysteine (Cys) residue6) (Fig. 2a). The third step begins with the formation of the S-acyl isoprotein 3 and is followed by the asparagine (Asn) side chain-assisted amide cleavage.7,8) The S-acylation can function as an amide cleavage signal in which the nitrogen lone-pair of the processed amide interacts with the π* orbital of the carbonyl group (Fig. 2b). As described below, such destabilization events observed in the intein system became the basis of our research.
3. Development of the N–S Acyl Transfer-Inducing Chemical Devices
3.1. Peptide Thioesters in Peptide SynthesisAs thioesters play a vital role in intein-mediated protein splicing, chemically synthesized peptide thioesters have had a tremendous impact on protein chemical synthesis through their application to native chemical ligation (NCL)9–12) (Fig. 3). The chemoselective reaction between the thioester 7 and an N-terminal Cys 8 as unprotected peptide is a feature in the NCL and a gold standard coupling protocol for protein synthesis. The NCL between 7 and 8 proceeds efficiently in an aqueous buffer solution to afford the NCL product 10 via the intermediate S-acyl isopeptide 9.
Much of protein chemical synthesis has taken advantage of the NCL protocol. However, two significant limitations are present. The first of these concerns the synthesis of peptide thioesters that have been prepared conventionally by solid-phase peptide synthesis (SPPS) based on t-butyloxycarbonyl (Boc) derivatives.13) Recently, peptide synthesis has moved from use of the Boc derivatives to 9-fluoroenylmethyloxycarbonyl (Fmoc)-based SPPS which involves treatment with base for the Fmoc removal step and, consequently, no Fmoc SPPS-compatible thioester-producing method existed14,15) (Fig. 4a(i)). The second limitation is that the conventional NCL protocol is incompatible with the sequential NCL coupling using more than two peptide fragments. This protocol is indispensable for the chemical synthesis of proteins with over 100 residues. The reason for its incompatibility is that the central fragment(s) require the N-terminal Cys and C-terminal thioester structure and thus risk suffering intramolecular NCL during the sequential ligation step (Fig. 4b).
The Fmoc procedure uses piperidine as a base for the Fmoc removal, and its application to the thioester-linked resin 11 leads to the racemization and decomposition of the thioester moiety in peptide fragments (Fig. 4a(i)). In this context, the Fmoc SPPS-mediated synthesis of thioesters requires conversion of an Fmoc-compatible amide (12 or 13) to an incompatible thioester 14 at the final stage of the synthesis16,17) (Fig. 4a(ii)).
3.2. Development of N-Sulfanylethylanilide (SEAlide) Unit Enabling N–S Acyl Transfer-Mediated Synthesis of Peptide ThioestersWe initially used a Cys-derived N-peptidyl oxazolidinone 15 to mimic the ground-state destabilization associated with the N–S acyl transfer step18) (Figs. 2a, 5). In this system, the electron-withdrawing ring carbonyl group inhibits the delocalization of the nitrogen lone-pair to the exo-amide carbonyl and contributes to twisting the amide bond. Subsequently, attack of the sulfenyl group on the destabilized amide converts the amide to the corresponding thioester 16. The use of the oxazolidinone system in the Fmoc SPPS led to the N–S acyl transfer-mediated synthesis of peptide thioesters. This system suffered however from the decomposition of the N-peptidyl oxazolidinone linkage and racemization during the peptide elongation step. Tuning the extent of destabilization of amide bonds was attempted by replacing the strong electron-withdrawing carbonyl group with an adjacent sp2 carbon unit, leading to an evaluation of the SEAlide unit. Peptides in which the SEAlide unit is incorporated, referred as to SEAlide peptides 17, can be easily synthesized by standard Fmoc SPPS.19,20)
Unlike the peptidyl oxazolidinone 15, the resulting amide-type SEAlide peptide 17 could not be easily converted to the corresponding thioester 18 under neutral conditions due to the less destabilized extent of its amide bond. Attempted N–S acyl transfer of 17 under neutral conditions failed, but under acidic conditions the reaction allowed the desired acyl transfer, yielding the peptide thioester 18. The observed results under both conditions can be explained by the difference in the trends of the energy diagram in Fig. 6.
3.3. Application of the SEAlide Unit in Protein SynthesisFollowing our initial observation, we attempted the NCL of a peptide thioester 19 with an N-terminal cysteinyl SEAlide peptide 20 under conventional NCL conditions in a neutral aqueous buffer containing sodium phosphate and guanidium hydrochloride (Gn·HCl). The formation was anticipated of a ligated amide-type SEAlide peptide 21 that can be converted by treatment with acid to the thioester, allowing the second NCL. We envisioned that the success in the attempted synthesis should pave the way to achievement of an N–to–C-directed sequential NCL. However, an unexpected cyclic peptide 22 derived from the intramolecular NCL of the N-terminal cysteinyl SEAlide thioester peptide 23 was formed in spite of the fact that no N–S transfer of the SEAlide unit occurred under neutral conditions20) (Fig. 7).
The observed and unexpected result moved us to examine whether the SEAlide unit can act as thioester under neutral conditions with the model ligation of 24 and 25. The attempted NCL clearly proceeded, affording the desired NCL product 28, and progress of this unexpected NCL was shown to require a phosphate salt as an additive in the reaction mixture21,22) (Fig. 8).
This phenomenon can be explained as follows: (1) The thermodynamically unfavored N–S acyl transfer (24 to 26) of the amide-type SEAlide peptide 24 occurs with thermodynamic compensation resulting from the subsequent conversion of the unstable thioesters (26 or 27) to the amide product 28 (Fig. 9a); (2) The phosphate salt will work as an acid-base catalyst for the N–S acyl transfer and lower the energy requirements for generation of the thioester 2623) (Fig. 9b). The trend of the overall energy diagram describing the unexpected NCL reaction is viewed as conceptually identical to that of intein-mediated protein splicing (Fig. 9c). Although the SEAlide unit was initially developed as an N–S acyl transfer device modeled on the first step of the intein system, the straightforward combination of the SEAlide system with NCL allows the partial mimicking the intein system from the viewpoint of the overall energy diagram.
This finding encouraged us to apply the SEAlide peptide to an N–to–C-directed one-pot/sequential NCL which allows efficient chemical access to >100 mer proteins using more than two peptide fragments. The envisioned one-pot/sequential NCL using the SEAlide peptide is shown in Fig. 10.21,24–27) The first NCL of peptide thioesters 19 with an N-terminal cysteinyl SEAlide peptide 20 in the absence of phosphate salts enables the SEAlide moiety to remain intact, and the amide-type ligated SEAlide peptide 21 can be obtained. Subsequently, addition of an N-terminal cysteinyl peptide 29 in phosphate buffer to the reaction mixture as a third component allows the resulting SEAlide peptide 21 to participate in the second NCL as a peptide thioester 30, leading to the formation of the fully ligated protein 31 in a one-pot/sequential manner. The developed one-pot/sequential NCL protocol using the SEAlide peptides represents one format of the N–to–C-directed methods. It has showcased its utility in protein chemical synthesis by its application to various proteins, including 77-residue CXCL14 chemokine28,29) and 162-residue glycosylated GM2-activator proteins (GM2AP)30–32) (Fig. 11).
In an attempt at synthesizing GM2AP, one-pot/sequential NCL of peptide fragments (34, 35 and 36) allowed the efficient preparation of the 88-mer C-half segment 38. Generally, HPLC purification of the ligation product accompanies the loss of desired material due to the unavoidable adsorption of peptides. Consequently, the one-pot/sequential operation which avoid purification of intermediary coupling products has been sought after, and our protocol should become a helpful method of choice for protein synthesis. Finally, the convergent SEAlide-mediated coupling of 38 with the glycosylated N-half segment 37 prepared from NCL of 32 with 33 yielded the desired biologically active glycosylated GM2AP after a refolding process. Such synthetic proteins have played indispensable roles in exploring the biological significance of these compounds.33–36) In particular, CXCL14-derived probe proteins are now in active use for identification of receptors, and the results obtained by our group will be reported in due course.
3.4. Application of SEAlide Units to Modification of a Targeting ProteinThe unique character of the SEAlide is that the N–S acyl transfer occurs in the presence of phosphate salts which work as an acid-base catalyst, and this provoked us to exploit the SEAlide moiety for a linker to modify target proteins37–39) (Fig. 12). Upon binding of the linker 39 to a target protein via a protein binder, or “bite,” the electrophilically inert SEAlide-containing linker connecting the protein binder as a bite and the biotin is activated, forming the corresponding thioester 40 which can acylate the ε-amino group of lysine located nearby the bite-binding pocket to produce a biotinylated protein 41. Concentration of the biotinylated protein using avidin beads followed by proteomic analyses of the biotinylated materials revealed information concerning the target proteins, including the identification of target itself and target binding sites.
4. Development of Protein Thioester-Producing Systems
4.1. Protein Thioesters in Protein ChemistryAs explained above, peptide thioesters have great utility in the science of peptides and proteins through their use in NCL, which leads to the synthesis of various proteins. The synthetic chemical approach using thioester-producing units such as SEAlide can afford peptide thioesters, but this synthetic approach is generally difficult to produce protein thioesters. The conversion of a naturally occurring peptide sequence 42 to a thioester should allow the preparation of the expressed protein-derived thioesters 43, and the resulting esters can turn into various useful proteins such as 45 through NCL with Cys-tagged artificial molecules 4440) (Fig. 13).
Consequently, several biochemical protocols to produce protein thioesters using intein41–45) or other enzymes46–48) have been published. Such biochemical protocols facilitate proteins with an intein- or enzyme recognition sites to be converted to the corresponding protein thioesters, but the defects in this approach are that the intein fusion proteins are less soluble and that enzyme recognition sites remain in the product in a non-traceless manner. In this context, we have engaged in the development of a chemistry-based methodology enabling the conversion of a naturally occurring peptide sequence to the corresponding thioester and have developed three distinct protocols.49)
4.2. Development of Sequential Quadruple Acyl Transfer (SQAT) SystemOne protocol uses a SQAT consisting of N–O, O–O, O–N and N–S transfers to produce thioesters50) (Fig. 14). Bal and colleagues reported that a serine (Ser)-Xaa-histidine (His)-Yaa sequence in proteins in which Xaa, Yaa are any amino acid could induce hydrolysis of the amide bond adjacent to the N-end of a Ser-Xaa-His-Yaa sequence upon adding Ni(II) salts to a solution of the protein (46 to 49).51,52) In this reaction, the coordination of Ni(II) to the peptide sequence shown in 47 inhibits the delocalization of the nitrogen lone pair of the peptidyl–Ser bond and destabilizes the amide bond. Subsequently, nucleophilic attack of the Ser hydroxyl group on the activated amide leads to the formation of O-peptidyl intermediate 48, which is then hydrolyzed to a protein acid 49. The intermediary-formed O-peptidyl intermediate 48 formed through the Ni(II)-mediated N–O acyl transfer attracted our attention due to the feasibility of its conversion to the peptide methyl esters 50 by the addition of MeOH to the Ni(II)-mediated reaction. Hydrazinolysis of the resulting peptide methyl ester 50 affords the peptide hydrazide 51 which can be converted to the peptide thioester 53 via the peptidyl azide 52 according to the protocol of Liu and colleagues.53,54) The SQAT protocol featuring the quadruple acyl transfers allowed the formation of peptide thioesters from the naturally occurring peptide sequence containing the Ser-Xaa-His-Yaa sequence. However, one problem is that the Ni(II)-mediated methanolysis requires a large excess amount of MeOH with its concentration in the reaction ranging from 10 to 50%, which impedes the broad application of the SQAT protocol to a wide variety of proteins due to the denaturation of the protein that is induced by MeOH.
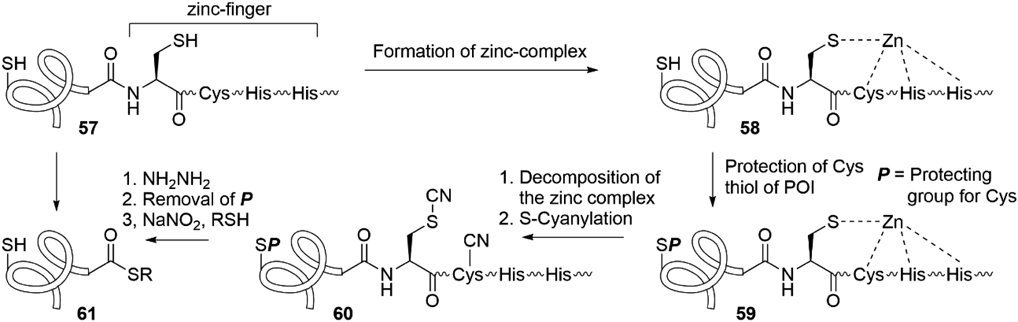
4.3. Development of an S-Cyanylation-Mediated Thioester-Producing SystemWe next explored a protocol that can be used under aqueous buffer conditions and attempted to apply the S-cyanylation-mediated amide hydrolysis reaction to the preparation of protein thioesters55) (Fig. 15). The amide bond of the S-cyanocysteine residue is susceptible to a nucleophile such as the H2O or NH4OH, and the cyanocysteine-containing peptides split into peptide acids or amides and 2-iminothiazolidine-4-carbonyl (Itc)-capped peptides.56–58) Such splitting could be explained by the hypothesis that the amide is destabilized by the inhibition of delocalization of the nitrogen lone pair by its interaction with the π* C≡N orbital (Fig. 15). The interaction of the amide nitrogen lone pair with a π* orbital, leading to destabilization of the amide, is also found in the final step of the intein-mediated protein splicing. This splicing involves amide cleavage triggered by the attack of Asn side chain and subsequent S–N acyl transfer (Figs. 2b, 15b). Based on the S-cyanylation-mediated hydrolysis, the reaction evolved into the production of peptide hydrazides as thioester precursors. The S-cyanylation of a peptide containing a single Cys 54 with 1-cyano-4-dimethylamino-pyridinium tetrafluoroborate,56) followed by treatment with hydrazine, afforded the non-Cys-containing peptide hydrazide 51 and the Itc-capped peptide 56. According to Liu’s protocol, the resulting hydrazide 51 could be converted to the peptide thioester 53 as shown in Fig. 15a. A drawback of this method is that its application to the preparation of Cys-containing thioesters requires the site-selective S-cyanylation of the cysteine located at the cleavage junction. This difficulty was overcome by using the fusion peptide 57 of a zinc-finger sequence at the C-terminal side of the peptide or the protein of interest (POI) (Fig. 16). Formation of the zinc-complex 58 allowed the synthesis of a site-selectively S-protected peptide 59, and the subsequent decomposition of the zinc complex provides an S-cyanylation site on the zinc-finger sequence. S-Cyanylation then gave a Cys-protected POI and a zinc-finger Cys-cyanylated peptide 60. The reaction of 60 with hydrazine proceeded with the release of the zinc finger sequence and afforded the S-protected POI hydrazide. Finally, removal of the S-protection followed by the conversion of the hydrazide to the corresponding thioester 61 allows the formation of peptide/protein thioester from the naturally occurring, Cys-containing sequences. This S-cyanylation-mediated protocol has found utility in the thioesterification of expressed proteins and has been used by other research groups.59–62)
4.4. Development of Thioester-Producing Chemoenzymatic Systems Enabled by Carboxypeptidase and Auto-processing ReactionsFollowing the cyanylation-mediated protocol, we developed a more sophisticated and versatile thioester-producing protocol using proteases.63) Of the various proteases, we focused on a carboxypeptidase, a type of exoprotease, because endopeptidases require the enzyme-recognition sequence for their use and following their non-traceless conversion remains challenging. We examined the applicability of carboxypeptidase Y (CPaseY),64) belonging to the serine protease family, to the thioester formation (Fig. 17). CPaseY is commercially available and can be easily isolated from dry yeast. It recognizes carboxylic acids at the C-terminus and releases the C-terminal amino acids upon hydrolysis of the intermediate O-acyl enzyme. Our initial hypothesis was that thiolysis of the O-acyl enzyme intermediate should lead to generation of the thioester. This attempt however failed and moved us to employ the hydrazinolysis strategy. Treatment of a model peptide with CPaseY in the presence of hydrazine undoubtedly gave the desired peptide hydrazide, which disappeared upon extension of the reaction due to the over-reaction by CPaseY.65) The temporary protection of the resulting hydrazide by hydrazone formation revealed a limited suppression of the over-reaction but failed to stop the further hydrolysis by CPaseY.
Therefore, we turned our attention to the substrate preference of CPaseY. CPaseY quickly releases hydrophobic C-terminal amino acids such as leucine (Leu), isoleucine (Ile), and valine (Val) whereas it hydrolyzes hydrophilic amino acids including arginine (Arg) and lysine (Lys), and proline (Pro) more slowly66) (Fig. 18a).
This substrate preference prompted us to utilize a two-residue C-terminus consisting of an unfavored–favored amino acid pair which suppress the over-reaction (Fig. 18b). Hydrazinolysis-mediated release of the C-terminal Leu from the Pro-Leu-tagged substrate peptide 62 proceeded efficiently without significant over-reaction and afforded the desired Pro hydrazide peptide 63. Among the residues that were examined, Pro was the best choice from the unfavored amino acids. The resulting Pro hydrazide 63 can be smoothly converted to the corresponding Pro thioester 64, but a problem that appeared was that the thioesters are limited to the Pro thioester which exhibits relatively low reactivity in NCL chemistry.67)
Incorporating an additional Cys residue at the N-terminus of the Pro overcame the problem (Fig. 19). The C-terminal Cys-Pro thioester unit was converted into the diketopiperazine-type thioester68,69) with the N–S acyl transfer involving the Cys thiol followed by diketopiperazine formation as is seen in the Cys-Pro-oxyester system.70,71) The CPaseY-mediated hydrazinolysis of the C-terminally Cys-Pro-Leu-tagged substrate 65 initially afforded the Pro hydrazide 66 that is converted to the corresponding thioester 67 by the standard protocol. Upon the conversion to the Pro thioester 67, spontaneous conversion to diketopiperazine-type thioester 69 occured through the N–S acyl transfer-mediated generation of an isopeptide intermediate 68 followed by diketopiperazine formation involving the Pro thioester. Finally, thiol exchange of 69 yielded thioesters 70 lacking the Cys-Pro-Leu tag sequence. The developed chemoenzymatic methodology enables conversion of the Cys-Pro-Leu-OH-tagged peptides 65 into peptide thioesters 70 in a traceless manner and the tag sequence disappears in this chemoenzymatic process. Application of the method to the expressed Cys-Pro-Leu-tagged glutathione S-transferase (GST) fusion protein, followed by NCL with an N-terminal cysteinyl biotinylated peptide, allowed the preparation of the artificial biotinylated GST protein.
A weak point of this protocol is that the optimal reaction conditions vary according to the amino acids adjacent to the Cys residue. The deep hydrophobic substrate binding pocket in CPaseY,72) a space that can tolerate a five-mer sequence, prefers to accept substrates possessing hydrophobic residues, such as 71 over those possessing the hydrophilic residues 72, which require an appropriate adjustment of the reaction time and amount of the enzyme, depending the residue on the N-side of the Cys (Fig. 20a). The incorporation of a repeat of the Cys-Pro sequence provably moves the residue affecting the reaction conditions out of the binding pocket. Consequently, the use of substrates (73 and 74) with three Cys-Pro three repeats allowed the normalization of the reaction conditions to afford the desired thioesters 77 with sequential release of Cys-Pro diketopiperazine units from the initially formed Pro thioester 76 generated from 7573) (Fig. 20b).
5. Development of Amide-Cleavage Systems
5.1. Development of Stimulus-Responsive Amide-Cleavage SystemsCleavage of amide bonds leads to a drastic change in protein function, and various enzymes that cleave a specific sequence have served as tools that can induce a functional change in a protein. In contrast to such a naturally occurring biochemical tool, the development of a chemical device triggering the amide cleavage remains a challenge. The trimethyl lock system,74–76) as a facile six-membered lactone-forming structure, moved us to apply the system to a stimulus-responsive amide cleavage amino acid 78.77–80) The sterically crowded nature of a neighborhood containing three methyl groups allows rapid lactonization involving the phenolic hydroxyl and neighboring carbonyl group with ester or amide cleavage occurring when X = OR or NHR, respectively (Fig. 21a). Based on this observation, we developed the amide cleavage dependent on removal of O-protection using 78 (Fig. 21b). The peptide 79 containing an appropriately protected 78 was split into two peptides (81 and 82) by removal of the protection (79 to 80). The half-lives of 79 depend on amino acids located at the N- and C-side of 78 and range from 3 min for Gly-78-His to 75 min for Ile-78-Tyr.81) The designed system features a selection in which the O-protection can control the amide cleavage without changing the mother structure of the amino acid82–84) (Fig. 21c). In addition to the trimethyl lock system, an intramolecular base-assisted system was developed (Fig. 22). The amide cleavage between the intein–C-extein junction accompanying the imide formation of the Asn residue requires a base catalyst to enhance the nucleophilicity of the side chain7,8) (Fig. 22a). We attempted to facilitate the imide formation by incorporating both a stimulus-regenerated basic unit and dimethyl group (Thorpe–Ingold effect) into the Asn side chain. The modified Asn-containing peptide 83 was proved to be converted into the imide peptide 85 via the intermediate 84 upon stimulus-responsive removal of the protection, although the trimethyl lock system proceeded more efficiently85) (Fig. 22b).
5.2. Application of an Amide Cleavage Device to Functional Change of PeptidesIn view of the final step of the intein-mediated protein splicing consisting of the amide cleavage followed by S–N acyl transfer (Fig. 1, 3 to 5 to 6) and O–N acyl transfer-mediated click peptide systems,86) we hypothesized that the application of the UV-responsive amide cleavage amino acid to an isopeptide system enabled the UV-dependent functional change of the peptide77,87) (Fig. 23). The parent peptide 86 connecting peptide A with an isopeptide B via an amide cleavable residue initially exhibits the function A by peptide A while the function B of peptide B is masked by the isopeptidyl format of peptide B. Upon UV irradiation, the removal of a photocleavable o-nitrobenzyl group (oNB) followed by amide cleavage, results in the formation of the O-acyl isopeptide B 87. The N–O peptidyl transfer immediately follows and affords the mature peptide B 88 that exhibits function B. This process should allow the functional change of peptides in a stimulus-responsive manner (Fig. 23a). Based on this hypothesis, we attempted to prepare shuttle77) and cellular protein-labeling peptides87) which possess the appropriate functional peptides A and B as shown in Fig. 23b.
There follows a brief description of the cellular protein-labeling peptide 89 that has a cell-penetrating peptide (CPP)88) and a split intein C-domain (IC)/fluorophore89) as functional peptides A and B, respectively.
The labeling peptide 89 initially moved into cells using the CPP function and then UV-irradiation-mediated removal of the oNB group induced the amide cleavage to yield the isopeptide format of split IC in the cells. The resulting isopeptide 90 immediately became mature IC 91 by O–N acyl transfer. Until this point, fluorescence quencher (Q) incorporated at the N-portion of the IC quenched fluorescence. Meanwhile, a split intein N-domain (IN)-fused protein of interest (POI) 92 was expressed in the cells where the N- and C-domain made an active intein complex 93 to exhibit intein activity. Subsequently, the split intein-mediated splicing allowed the coupling of the POI with fluorophore. This splicing process triggered the release of the quencher to yield the fluorescence POI 94 (Fig. 24).
5.3. Application of an Amide Cleavage Device to Target Protein IdentificationThe developed amide cleavage device 78 can cleave not only amide bonds but also acyl aminooxy bonds. The resulting aminooxy group, absent from biological samples, selectively reacts with aldehydes to afford stable oxime adducts. A traceable linker 95 enabling the selective visualization of a target protein from a complex proteome mixture was developed using 7890,91) (Fig. 25). The developed linker 95 has biotin and azide moieties, connected by the acyl aminooxymethyl unit, at both termini (Fig. 25).
One general strategy for identifying the target protein 96 of a ligand begins with the photoaffinity-based92) or activity-based93) labeling-mediated incorporation of alkyne units to the target (96 to 97). The resulting alkyne-presented target 97 is followed by the linking of the azide- and biotin-possessing linker by the copper-catalyzed azide-alkyne cycloaddition (CuAAC).94) Such an initial step allows the preparation of a biotin-presented target protein 98 that is then concentrated on the streptavidin (SAv) beads. Elution of the target from the SAv beads under the biotin-avidin dissociation conditions could lead to the identification of the target protein but the release of non-specific binders on the SAv beads sometimes prevents the precise identification of the target. The use of a cleavable linker enabling the selective elution of the biotin-presented target solves this elution-associated problem.95) We developed a traceable linker 9590) as an advanced cleavable linker, featuring a cleavable acyl aminooxy unit. Upon cleavage of the acyl aminooxy linkage by removal of the protection of the phenolic OH, the aminooxy group-bearing target protein 99 was eluted. The fact that no aminooxy group exists in biological samples allows the selective labeling of 99 with an aldehyde-containing reporter from a complex mixture, and only the labeled sample 100 was visualized. This process enables the highly selective visualization of target proteins from complex biological samples. However, in the case of low molecular weight targets, added reporters interfered with the visualization of targets in the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis.
One solution to this limitation used the aforementioned N–S acyl transfer system.37) As a more facile SEAlide-type acyl transfer unit, we developed the N-sulfanylethylcoumarinyl amide (SECmide)96) that is incorporated into an on/off fluorescent traceable linker 10197) (Fig. 26). Concentration of an alkyne-presented target 97 on the SAv beads using 101 was performed by a protocol identical to that using 95. Subjection of the target-immobilized beads 102 to an NCL reaction with cysteine induced the N–S acyl transfer at the N-acyl SECmide unit (102 to 103) and allowed the elution of the non-N-acylated SECmide-bearing target 104 from the SAv beads. The N-acylated 7-aminocoumarin exhibits no fluorescence while the corresponding non-N-acylated form shows fluorescence. Therefore, the NCL-mediated elution enabled the selective visualization of a target by means of the fluorescent signal.
6. Miscellaneous
6.1. Synthesis of Fluoroalkene-Dipeptide Isosteres Inspired by Acetyl-CoA-Generating ReactionAs described above, the intein-mediated protein-editing system has provided various clues to advance peptide/protein chemistry. In addition to the topics presented here, we undertook an effort to develop a synthetic method for thioesters based on the acetyl-CoA generating system. In this system, the thiamin diphosphate-derived N-heterocyclic carbene 105 (NHC)-mediated intermolecular redox reaction of pyruvate 106 (a reductant) with lipoamide 107 (an oxidant) leads to the formation of acetyl-CoA 108 (Fig. 27a).
However, the failure in mimicking this system for producing peptide thioesters moved our attention to its application to the synthesis of fluoroalkene dipeptide isosteres 109 as stable dipeptide biomimetics. We have developed the synthetic methodology for the isosteres that use organocopper- or SmI2-mediated reduction of γ,γ-difluoro-α,β-enoate 11098–100) (Fig. 27b). Successive single electron transfers (SET) from the reagents to the substrate complete the reaction. Based on this mechanism, a hypothesis that the NHC-mediated intramolecular redox reaction can apply to the preparation of the isostere was formulated. Consequently, application of the NHC-mediated reaction to the intramolecular redox reaction of a γ,γ-difluoro-α,β-enoylsilane system 111 allowed the synthesis of the fluoroalkene dipeptide isostere 109 as a hydrolytically stable dipeptide mimetic, in which the acylsilane unit intramolecularly reduced the allylic difluoride in an SN2′ fashion with accompanying defluorination101,102) (Fig. 27c).
6.2. Rediscovery of the Chemistry of S-Protected Cysteine Sulfoxide Triggered by Protein SynthesisIn addition to the N–to–C-directed sequential NCL protocol using a cryptic thioester such as the SEAlide unit (Figs. 10, 11, 28a), the C–to–N-ligation utilizing the thiazolidine-4-carbonyl (Thz) peptide thioester 112 as a central fragment also represents another sequential ligation method103) (Fig. 28b). Thz protection is also indispensable for the N–to–C-directed preparation of a large C-half segment for convergent coupling strategy as shown in the synthesis of GM2AP (Fig. 11). In this context, we explored a Thz-opening reaction as an alternative to the conventional ring-opening reaction using O-methylhydroxylamine (Fig. 28c). We found that a Cu(II) salt acts as a Lewis acid and opens the ring104) (Fig. 28d), and this has led to new chemistry of peptide modification. This began with the unexpected finding that the treatment of a Thz- and S-acetamidomethyl Cys (Cys(Acm))-containing peptide 113 gives the corresponding disulfide peptide 114.105) We hypothesized that the initially generated Cys–peptide 115 is converted to S-chlorocysteine (Cys(Cl))-containing peptide 117 via a Cys sulfenic acid (Cys(OH)) peptide 116 by the reaction with Cu(II) as an oxidant and chloride anion, and the resulting product 117 is converted to the disulfide peptide 114 by the nucleophilic attack of the sulfide of the Cys(Acm) on the electrophilic Cys(Cl) (Fig. 29a). Similarity, the indole of tryptophan attacked the Cys(Cl) derived from the Thz residue and then aromatic electrophilic substitution (SNAr) occurred to give tryptathionine moiety (118 to 120)106) (Fig. 29b).
Reactions including disulfide bond and tryptathionine formation require the oxidative conversion of Cys to Cys(Cl). Such an oxidation generally proceeds smoothly only at the N-terminal Cys; however, oxidation of the internal Cys residue to the corresponding Cys(Cl) requires harsh conditions which cause side reactions on oxidation sensitive amino acid residues. We envisioned that oxidation-free generation of the Cys(Cl) in peptides should enhance the synthetic usefulness of the reactions involving Cys(Cl). A clue to advancing this idea came from our research group which has used S-protected Cys sulfoxides for about 30 years.107–109) We realized that S-protected Cys sulfoxides could be converted to the corresponding Cys(Cl) unit under acidic conditions in the presence of chloride anion in a redox-neutral manner. Under these conditions, S-p-methoxybenzyl Cys sulfoxide (Cys(MBzl)(O)) reacted efficiently with the Trp in peptide 121 to afford the tryptathionine-containing peptide 123 via an intermediate 122110) (Fig. 30), and the success led to late-stage peptide lipidation.111) In addition, regioselective disulfide formation using S-protected Cys sulfoxides has become available. Further research efforts for applying the sulfoxide chemistry to peptide/protein science, including late-stage peptide/protein modification and syntheses of peptides containing multiple disulfides and side chain-bridged peptides, have continued in our group.
7. Conclusion
This account has described our efforts to advance peptide/protein chemistry. Naturally occurring events associated with biomolecules, including proteins in physiological environments, have served as our textbook with which to achieve our research. Chemically mimicking the intein-mediated protein-editing system with its sequential acyl transfers has provided an indispensable scientific background for performing research and has led to the development of the N–S acyl transfer-mediated thioester-producing device termed SEAlide, a protein thioester-generating protocol operating in a traceless manner and a stimulus-responsive amide cleavage amino acid with applications in chemical biology field. In particular, the use of the SEAlide in NCL protocols enabled the unprecedented N–to–C-directed one-pot/sequential NCL coupling regulated by acid-base catalysts such as phosphate salts. This has permitted the efficient chemical synthesis of proteins with over 150 residues. The NHC-mediated generation of acetyl-CoA also is a sterling example which has achieved the synthesis of fluoroalkene dipeptide isosteres. Our efforts to synthesize large proteins fortuitously rediscovered the S-protected Cys sulfoxide chemistry that has remained unnoticed even though our group found it over 30 years ago. The rediscovered chemistry has now been included in an active and unprecedented application to peptide/protein chemistry.
Acknowledgments
I sincerely thank the dedicated collaborators in my group. Their enthusiastic contributions, both intellectually and experimentally, made this research possible.
This research was supported by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) and by Grants by the Suzuki Memorial Foundation, Novartis Foundation, Nagase Science and Technology Foundation, the Uehara Memorial Foundation, and the Canon Foundation and “Innovation inspired by Nature” Research Support Program (Sekisui Chemical Co., Ltd.).
Conflict of Interest
The author declares no conflict of interest.
Notes
This review of the author’s work was written by the author upon receiving the 2022 Pharmaceutical Society of Japan Award.
References
- 1) Shih C. K., Wagner R., Feinstein S., Kanik-Ennulat C., Neff N., Mol. Cell. Biol., 8, 3094–3103 (1988).
- 2) Hirata R., Ohsumk Y., Nakano A., Kawasaki H., Suzuki K., Anraku Y., J. Biol. Chem., 265, 6726–6733 (1990).
- 3) Kane P. M., Yamashiro C. T., Wolczyk D. F., Neff N., Goebl M., Stevens T. H., Science, 250, 651–657 (1990).
- 4) Vila-Perello M., Muir T. W., Cell, 143, 191–200 (2010).
- 5) Mills K. V., Johnson M. A., Perler F. B., J. Biol. Chem., 289, 14498–14505 (2014).
- 6) Romanelli A., Shekhtman A., Cowburn D., Muir T. W., Proc. Natl. Acad. Sci. U.S.A., 101, 6397–6402 (2004).
- 7) Shemella P., Pereira B., Zhang Y., Van Roey P., Belfort G., Garde S., Nayak S. K., Biophys. J., 92, 847–853 (2007).
- 8) Frutos S., Goger M., Giovani B., Cowburn D., Muir T. W., Nat. Chem. Biol., 6, 527–533 (2010).
- 9) Dawson P., Muir T., Clark-Lewis I., Kent S., Science, 266, 776–779 (1994).
- 10) Dawson P. E., Kent S. B. H., Annu. Rev. Biochem., 69, 923–960 (2000).
- 11) Hackenberger C. P. R., Schwarzer D., Angew. Chem. Int. Ed., 47, 10030–10074 (2008).
- 12) Agouridas V., El Mahdi O., Diemer V., Cargoët M., Monbaliu J.-C. M., Melnyk O., Chem. Rev., 119, 7328–7443 (2019).
- 13) Hironobu H., Saburo A., Bull. Chem. Soc. Jpn., 64, 111–117 (1991).
- 14) Kang J., Macmillan D., Org. Biomol. Chem., 8, 1993–2002 (2010).
- 15) Mende F., Seitz O., Angew. Chem. Int. Ed., 50, 1232–1240 (2011).
- 16) Hojo H., Onuma Y., Akimoto Y., Nakahara Y., Nakahara Y., Tetrahedron Lett., 48, 25–28 (2007).
- 17) Dheur J., Ollivier N., Vallin A., Melnyk O., J. Org. Chem., 76, 3194–3202 (2011).
- 18) Ohta Y., Itoh S., Shigenaga A., Shintaku S., Fujii N., Otaka A., Org. Lett., 8, 467–470 (2006).
- 19) Tsuda S., Shigenaga A., Bando K., Otaka A., Org. Lett., 11, 823–826 (2009).
- 20) Sakamoto K., Sato K., Shigenaga A., Tsuji K., Tsuda S., Hibino H., Nishiuchi Y., Otaka A., J. Org. Chem., 77, 6948–6958 (2012).
- 21) Sato K., Shigenaga A., Tsuji K., Tsuda S., Sumikawa Y., Sakamoto K., Otaka A., ChemBioChem, 12, 1840–1844 (2011).
- 22) Aihara K., Inokuma T., Jichu T., Lin Z., Fu F., Yamaoka K., Shigenaga A., Hutchins D. A., Schmidt E. W., Otaka A., Synlett, 28, 1944–1949 (2017).
- 23) Shigenaga A., Org. Biomol. Chem., 18, 9706–9711 (2020).
- 24) Ding H., Shigenaga A., Sato K., Morishita K., Otaka A., Org. Lett., 13, 5588–5591 (2011).
- 25) Otaka A., Sato K., Ding H., Shigenaga A., Chem. Rec., 12, 479–490 (2012).
- 26) Aihara K., Yamaoka K., Naruse N., Inokuma T., Shigenaga A., Otaka A., Org. Lett., 18, 596–599 (2016).
- 27) Otaka A., Sato K., Shigenaga A., Chemical Synthesis of Proteins Using N–Sulfanylethylanilide Peptides, Based on N–S Acyl Transfer Chemistry. “Protein Ligation and Total Synthesis II,” ed. by Liu L., Topics in Current Chemistry 363, Springer, Switzerland, 2015, pp 33–56.
- 28) Tsuji K., Shigenaga A., Sumikawa Y., Tanegashima K., Sato K., Aihara K., Hara T., Otaka A., Bioorg. Med. Chem., 19, 4014–4020 (2011).
- 29) Tsuji K., Tanegashima K., Sato K., Sakamoto K., Shigenaga A., Inokuma T., Hara T., Otaka A., Bioorg. Med. Chem., 23, 5909–5914 (2015).
- 30) Sato K., Shigenaga A., Kitakaze K., Sakamoto K., Tsuji D., Itoh K., Otaka A., Angew. Chem. Int. Ed., 52, 7855–7859 (2013).
- 31) Sato K., Kitakaze K., Nakamura T., Naruse N., Aihara K., Shigenaga A., Inokuma T., Tsuji D., Itoh K., Otaka A., Chem. Commun., 51, 9946–9948 (2015).
- 32) Nakamura T., Sato K., Naruse N., Kitakaze K., Inokuma T., Hirokawa T., Shigenaga A., Itoh K., Otaka A., ChemBioChem, 17, 1986–1992 (2016).
- 33) Tanegashima K., Suzuki K., Nakayama Y., Tsuji K., Shigenaga A., Otaka A., Hara T., FEBS Lett., 587, 1731–1735 (2013).
- 34) Tanegashima K., Tsuji K., Suzuki K., Shigenaga A., Otaka A., Hara T., FEBS Lett., 587, 3770–3775 (2013).
- 35) Tanegashima K., Takahashi R., Nuriya H., Iwase R., Naruse N., Tsuji K., Shigenaga A., Otaka A., Hara T., EBioMedicine, 24, 247–256 (2017).
- 36) Kitakaze K., Mizutani Y., Sugiyama E., Tasaki C., Tsuji D., Maita N., Hirokawa T., Asanuma D., Kamiya M., Sato K., Setou M., Urano Y., Togawa T., Otaka A., Sakuraba H., Itoh K., J. Clin. Invest., 126, 1691–1703 (2016).
- 37) Morisaki T., Denda M., Yamamoto J., Tsuji D., Inokuma T., Itoh K., Shigenaga A., Otaka A., Chem. Commun., 52, 6911–6913 (2016).
- 38) Denda M., Morisaki T., Kohiki T., Yamamoto J., Sato K., Sagawa I., Inokuma T., Sato Y., Yamauchi A., Shigenaga A., Otaka A., Org. Biomol. Chem., 14, 6244–6251 (2016).
- 39) Kohiki T., Kato Y., Nishikawa Y., Yorita K., Sagawa I., Denda M., Inokuma T., Shigenaga A., Fukui K., Otaka A., Org. Biomol. Chem., 15, 5289–5297 (2017).
- 40) Thompson R. E., Muir T. W., Chem. Rev., 120, 3051–3126 (2020).
- 41) Muir T. W., Sondhi D., Cole P. A., Proc. Natl. Acad. Sci. U.S.A., 95, 6705–6710 (1998).
- 42) Evans T. C. Jr., Benner J., Xu M.-Q., Protein Sci., 7, 2256–2264 (1998).
- 43) Flavell R. R., Muir T. W., Acc. Chem. Res., 42, 107–116 (2009).
- 44) Shah N. H., Muir T. W., Chem. Sci., 5, 446–461 (2014).
- 45) Conibear A. C., Watson E. E., Payne R. J., Becker C. F. W., Chem. Soc. Rev., 47, 9046–9068 (2018).
- 46) Ling J. J., Policarpo R. L., Rabideau A. E., Liao X., Pentelute B. L., J. Am. Chem. Soc., 134, 10749–10752 (2012).
- 47) Cao Y., Nguyen G. K. T., Tam J. P., Liu C.-F., Chem. Commun., 51, 17289–17292 (2015).
- 48) Morgan H. E., Turnbull W. B., Webb M. E., Chem. Soc. Rev., 51, 4121–4145 (2022).
- 49) Denda M., Otaka A., Chem. Pharm. Bull., 70, 316–323 (2022).
- 50) Tsuda Y., Shigenaga A., Tsuji K., Denda M., Sato K., Kitakaze K., Nakamura T., Inokuma T., Itoh K., Otaka A., ChemistryOpen, 4, 448–452 (2015).
- 51) Krȩżel A., Kopera E., Protas A. M., Poznański J., Wysłouch-Cieszyńska A., Bal W., J. Am. Chem. Soc., 132, 3355–3366 (2010).
- 52) Kopera E., Krȩżel A., Protas A. M., Belczyk A., Bonna A., Wysłouch-Cieszyńska A., Poznański J., Bal W., Inorg. Chem., 49, 6636–6645 (2010).
- 53) Fang G.-M., Li Y.-M., Shen F., Huang Y.-C., Li J.-B., Lin Y., Cui H.-K., Liu L., Angew. Chem. Int. Ed., 50, 7645–7649 (2011).
- 54) Fang G.-M., Wang J.-X., Liu L., Angew. Chem. Int. Ed., 51, 10347–10350 (2012).
- 55) Miyajima R., Tsuda Y., Inokuma T., Shigenaga A., Imanishi M., Futaki S., Otaka A., Biopolymers, 106, 531–546 (2016).
- 56) Wakselman M., Guibé-Jampel E., Raoult A., Busse W. D., J. Chem. Soc. Chem. Commun., 21–22 (1976).
- 57) Nakagawa S., Tamakashi Y., Hamana T., Kawase M., Taketomi S., Ishibashi Y., Nishimura O., Fukuda T., J. Am. Chem. Soc., 116, 5513–5514 (1994).
- 58) Otaka A., 1-Cyano-4-Dimethylaminopyridinium Tetrafluoroborate. “Encyclopedia of Reagents for Organic Synthesis”: ‹https://doi.org/10.1002/047084289X.rn02408›
- 59) Kajihara Y., Kanemitsu Y., Nishihara M., Okamoto R., Izumi M., J. Pept. Sci., 20, 958–963 (2014).
- 60) Qiao Y., Yu G., Kratch K. C., Wang X. A., Wang W. W., Leeuwon S. Z., Xu S., Morse J. S., Liu W. R., J. Am. Chem. Soc., 142, 7047–7054 (2020).
- 61) Li Q.-Q., Liu Y.-Q., Luo Y.-Y., Chu T.-T., Gao N., Chen P.-G., Chen Y.-X., Li Y.-M., Chem. Commun., 56, 5370–5373 (2020).
- 62) Mo Z., Lin S., Chen W., He C., Angew. Chem. Int. Ed., 61, e202115377 (2022).
- 63) Komiya C., Shigenaga A., Tsukimoto J., Ueda M., Morisaki T., Inokuma T., Itoh K., Otaka A., Chem. Commun., 55, 7029–7032 (2019).
- 64) Endrizzi J. A., Breddam K., Remington S. J., Biochemistry, 33, 11106–11120 (1994).
- 65) Hamberg A., Kempka M., Sjödahl J., Roeraade J., Hult K., Anal. Biochem., 357, 167–172 (2006).
- 66) Hayashi R., Bai Y., Hata T., J. Biochem., 77, 69–79 (1975).
- 67) Pollock S. B., Kent S. B. H., Chem. Commun., 47, 2342–2344 (2011).
- 68) Raibaut L., Seeberger P., Melnyk O., Org. Lett., 15, 5516–5519 (2013).
- 69) Nakamura T., Shigenaga A., Sato K., Tsuda Y., Sakamoto K., Otaka A., Chem. Commun., 50, 58–60 (2014).
- 70) Kawakami T., Aimoto S., Tetrahedron, 65, 3871–3877 (2009).
- 71) Kawakami T., Shimizu S., Aimoto S., Bull. Chem. Soc. Jpn., 83, 570–574 (2010).
- 72) Nakase H., Murata S., Ueno H., Hayashi R., Biosci. Biotechnol. Biochem., 65, 2465–2471 (2001).
- 73) Ueda M., Komiya C., Arii S., Kusumoto K., Denda M., Okuhira K., Shigenaga A., Otaka A., Chem. Pharm. Bull., 68, 1226–1232 (2020).
- 74) Milstien S., Cohen L. A., Proc. Natl. Acad. Sci. U.S.A., 67, 1143–1147 (1970).
- 75) Jung M. E., Piizzi G., Chem. Rev., 105, 1735–1766 (2005).
- 76) Levine M. N., Raines R. T., Chem. Sci., 3, 2412–2420 (2012).
- 77) Shigenaga A., Tsuji D., Nishioka N., Tsuda S., Itoh K., Otaka A., ChemBioChem, 8, 1929–1931 (2007).
- 78) Shigenaga A., Yamamoto J., Nishioka N., Otaka A., Tetrahedron, 66, 7367–7372 (2010).
- 79) Shigenaga A., Yamamoto J., Kohiki T., Inokuma T., Otaka A., J. Pept. Sci., 23, 505–513 (2017).
- 80) Shigenaga A., Chem. Pharm. Bull., 67, 1171–1178 (2019).
- 81) Shigenaga A., Yamamoto J., Hirakawa H., Yamaguchi K., Otaka A., Tetrahedron, 65, 2212–2216 (2009).
- 82) Shigenaga A., Yamamoto J., Hirakawa H., Ogura K., Maeda N., Morishita K., Otaka A., Tetrahedron Lett., 51, 2525–2528 (2010).
- 83) Shigenaga A., Yamamoto J., Sumikawa Y., Furuta T., Otaka A., Tetrahedron Lett., 51, 2868–2871 (2010).
- 84) Shigenaga A., Ogura K., Hirakawa H., Yamamoto J., Ebisuno K., Miyamoto L., Ishizawa K., Tsuchiya K., Otaka A., ChemBioChem, 13, 968–971 (2012).
- 85) Komiya C., Aihara K., Morishita K., Ding H., Inokuma T., Shigenaga A., Otaka A., J. Org. Chem., 81, 699–707 (2016).
- 86) Taniguchi A., Sohma Y., Kimura M., Okada T., Ikeda K., Hayashi Y., Kimura T., Hirota S., Matsuzaki K., Kiso Y., J. Am. Chem. Soc., 128, 696–697 (2006).
- 87) Jung D., Sato K., Min K., Shigenaga A., Jung J., Otaka A., Kwon Y., Chem. Commun., 51, 9670–9673 (2015).
- 88) Futaki S., Adv. Drug Deliv. Rev., 57, 547–558 (2005).
- 89) Li Y., Biotechnol. Lett., 37, 2121–2137 (2015).
- 90) Yamamoto J., Denda M., Maeda N., Kita M., Komiya C., Tanaka T., Nomura W., Tamamura H., Sato Y., Yamauchi A., Shigenaga A., Otaka A., Org. Biomol. Chem., 12, 3821–3826 (2014).
- 91) Yamamoto J., Maeda N., Komiya C., Tanaka T., Denda M., Ebisuno K., Nomura W., Tamamura H., Sato Y., Yamauchi A., Shigenaga A., Otaka A., Tetrahedron, 70, 5122–5127 (2014).
- 92) Lapinsky D. J., Bioorg. Med. Chem., 20, 6237–6247 (2012).
- 93) Li N., Overkleeft H. S., Florea B. I., Curr. Opin. Chem. Biol., 16, 227–233 (2012).
- 94) Hein J. E., Fokin V. V., Chem. Soc. Rev., 39, 1302–1315 (2010).
- 95) Bielski R., Witczak Z., Chem. Rev., 113, 2205–2243 (2013).
- 96) Eto M., Naruse N., Morimoto K., Yamaoka K., Sato K., Tsuji K., Inokuma T., Shigenaga A., Otaka A., Org. Lett., 18, 4416–4419 (2016).
- 97) Morisaki T., Shigenaga A., Otaka A., Chem. Pharm. Bull., 68, 216–219 (2020).
- 98) Otaka A., Watanabe H., Mitsuyama E., Yukimasa A., Tamamura H., Fujii N., Tetrahedron Lett., 42, 285–287 (2001).
- 99) Otaka A., Watanabe H., Yukimasa A., Oishi S., Tamamura H., Fujii N., Tetrahedron Lett., 42, 5443–5446 (2001).
- 100) Otaka A., Watanabe J., Yukimasa A., Sasaki Y., Watanabe H., Kinoshita T., Oishi S., Tamamura H., Fujii N., J. Org. Chem., 69, 1634–1645 (2004).
- 101) Yamaki Y., Shigenaga A., Tomita K., Narumi T., Fujii N., Otaka A., J. Org. Chem., 74, 3272–3277 (2009).
- 102) Yamaki Y., Shigenaga A., Li J., Shimohigashi Y., Otaka A., J. Org. Chem., 74, 3278–3285 (2009).
- 103) Bang D., Kent S. B. H., Angew. Chem. Int. Ed., 43, 2534–2538 (2004).
- 104) Naruse N., Kobayashi D., Ohkawachi K., Shigenaga A., Otaka A., J. Org. Chem., 85, 1425–1433 (2020).
- 105) Kobayashi D., Naruse N., Denda M., Shigenaga A., Otaka A., Org. Biomol. Chem., 18, 8638–8645 (2020).
- 106) Kobayashi D., Kohmura Y., Hayashi J., Denda M., Tsuchiya K., Otaka A., Chem. Commun., 57, 10763–10766 (2021).
- 107) Funakoshi S., Fujii N., Akaji K., Irie H., Yajima H., Chem. Pharm. Bull., 27, 2151–2156 (1979).
- 108) Fujii N., Otaka A., Watanabe T., Arai H., Funakoshi S., Bessho K., Yajima H., J. Chem. Soc. Chem. Commun., 1987, 1676–1678 (1987).
- 109) Fujii N., Otaka A., Funakoshi S., Watanabe T., Arai H., Bessho K., Yajima H., J. Protein Chem., 7, 151–156 (1988).
- 110) Kobayashi D., Kohmura Y., Sugiki T., Kuraoka E., Denda M., Fujiwara T., Otaka A., Chem. Eur. J., 27, 14092–14099 (2021).
- 111) Kobayashi D., Kuraoka E., Hayashi J., Yasuda T., Kohmura Y., Denda M., Harada N., Inagaki N., Otaka A., ACS Med. Chem. Lett., 13, 1125–1130 (2022).