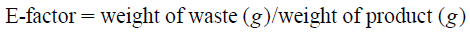Abstract
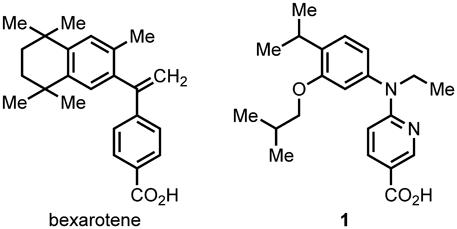
Small-molecular drugs, which are generally inexpensive compared with biopharmaceuticals and can often be taken orally, may contribute to the Sustainable Development Goals (SDGs) adopted by the United Nations. We previously reported the retinoid X receptor (RXR) agonist 4-(ethyl(3-isobutoxy-4-isopropylphenyl)amino)benzoic acid (NEt-3IB, 1) as a small-molecular drug candidate to replace biopharmaceuticals for the treatment of inflammatory bowel disease. The previous synthetic method to 1 required a large amount of organic solvent and extensive purification. In line with the SDGs, we aimed to develop an environmentally friendly, inexpensive method for the large-scale synthesis of 1. The developed method requires only a hydrophobic ether and EtOH as reaction and extraction solvents. The product was purified by recrystallization twice to afford 99% pure 1 at 100 mmol scale in about 30% yield. The optimized process showed a 35-fold improvement of the E-factor (an index of environmental impact) compared to the original method. This work, which changes the solvent used to environmentally preferable ones based on the existing synthetic method for 1, illustrates how synthetic methods for small-molecular drugs can be adapted and improved to contribute to the SDGs.
Introduction
In 2015, the United Nations (UN) Summit listed 17 Sustainable Development Goals (SDGs) to be achieved in the 15 years to 2030.1) One of them, “Ensure healthy lives and promote well-being for all at all ages (SDG 3)” was established partly due to the disparity in drug supply between developed and developing countries.2) In recent years, various biopharmaceuticals have been developed to treat intractable diseases, such as inflixivab and adalibumab for the treatment of inflammatory bowel disease (IBD). However, the efficacy of biopharmaceuticals can be compromised by the emergence of antibodies, and they are generally expensive to produce. From this point of view, small-molecular drugs are again becoming attractive.
Small-molecular drugs are generally relatively inexpensive and can often be taken orally, but on the other hand, they have disadvantages such as a greater likelihood of off-target effects compared to biopharmaceuticals. Current small-molecular drugs used to treat IBD include 5-aminosalycylic acid and its derivatives, steroidal anti-inflammatory drugs, the Janus kinase (JAK) inhibitor tofacitinib, and the immunosuppressant azathioprine, but these have varying efficacy for mucosal healing and remission,3) and there is still a need for alternatives.
We focused on retinoid X receptor (RXR) as a target to treat IBDs. The RXR agonist bexarotene (Targretin®, Fig. 1) has been used to treat cutaneous T-cell lymphoma (CTCL), and has been the subject of drug repositioning studies for various inflammatory diseases, including IBD.4–7) However, bexarotene causes side effects such as hypothyroidism.8,9) Therefore, we aimed to create RXR ligands with reduced side effects. We previously found that 4-(ethyl(3-isobutoxy-4-isopropylphenyl)amino)benzoic acid10) (NEt-3IB, 1, Fig. 1) maintains the ability to activate RXR transcription without causing the above side effects.11) Positron emission tomography (PET) imaging revealed that 1 accumulates in the liver after administration and is rapidly transferred to the lower gastrointestinal tract.12) A therapeutic effect of 1 accompanied with mucosal healing was confirmed in IBD model mice.13)
To obtain sufficient amounts of 1 for further evaluation, we required a scaled-up, low-cost, green synthetic process. In particular, the key objective was to minimize the use of organic solvents in line with the UN’s SDGs, such as “Ensure availability and sustainable management of water and sanitation for all (SDG 6).” However, our original synthesis of 1 uses various organic solvents and requires multiple purifications by column chromatography10) (Chart 1, Medicinal Chemistry method). Therefore, in this study, we focused on hydrophobic ether solvents that are widely used in process chemistry, not only as reaction media, but also as extraction solvents. For process chemistry, cyclopenthylmethyl ester (CPME), 2-methyltetrahydrofuran (2-MeTHF), 4-methyltetrahydropyran (MTHP) were considered as candidates.14–17) These solvents can be azeotropically dehydrated and reused after reactions, so that organic solvent waste is minimized. In this study, we aimed to establish a new process synthesis of 1 using CPME as an organic solvent, since it is inexpensive and readily available. The finally developed method required only CPME and EtOH. The product was obtained in a total of 7 steps, and purified by recrystallization twice, affording 99% pure 1 at 100 mmol scale in at least 30% overall yield.
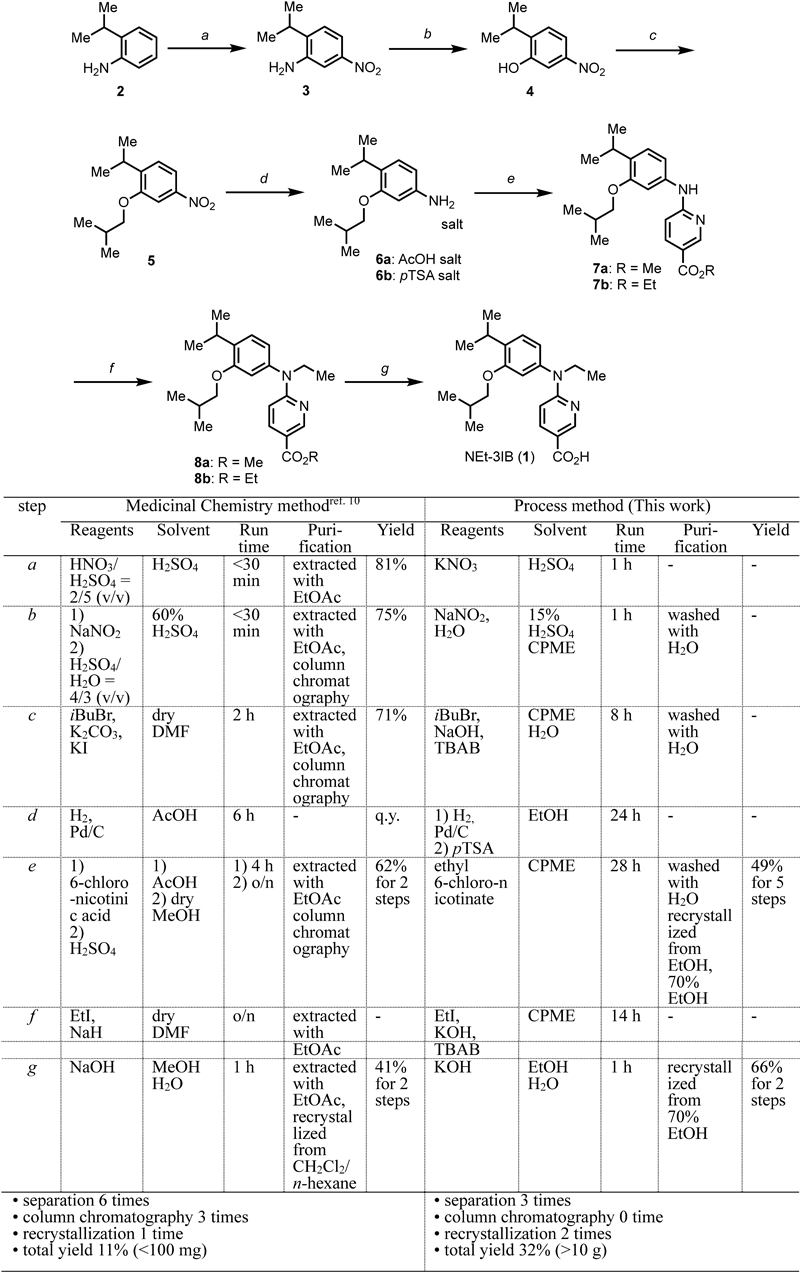
Chart 1. The Medicinal Chemistry Method and the Newly Developed Process Method to 1
Results and Discussion
Examination of Conditions for Each Reaction StepStep a: Nitration
The first step is the nitration of 2-isopropylaniline (2). In the Medicinal Chemistry method, nitroaniline 3 was synthesized using mixed acid (HNO3/H2SO4 = 2/5).10) However, in increasing the reaction scale, the use of a highly reactive mixed acid may be dangerous, and nitric acid is volatile, so that it is difficult to calculate the number of equivalents precisely. Therefore, KNO3 was added as a nitrating reagent in this step. The solubility of 2 was 3 mmol in 600 µL of conc.H2SO4, and so this was used as the maximum concentration for examination in Step b. It was considered that the nitroaniline 3 can be obtained as a single product by adding 1 eq of KNO3 to a conc.H2SO4 solution of 2.
Step b: Sandmeyer reaction
In the Medicinal Chemistry method, after confirming the completion of Step a, neutralization, extraction with EtOAc, and evaporation under reduced pressure were performed, and the residue was dissolved in dil.H2SO4 again to carry out Step b.10) In order to prevent solvent wastage due to the neutralization and extraction, we considered that Step b should be performed continuously by adding the conc.H2SO4 solution of nitroaniline 3 to ice to obtain dil.H2SO4 solution. In the Medicinal Chemistry method, nitroaniline 3 was suspended in dil.H2SO4, and NaNO2aq was added dropwise to form the diazonium salt, and then the solution was then added to heated dil.H2SO4 (about 50%) to obtain nitrophenol 4.10) To avoid the increase in the volume of the reaction solution, NaNO2 was added as a powder to the cooled dil.H2SO4 solution of nitrophenol 4. In addition, we considered that dropping the diazonium salt solution into heated dil.H2SO4 should be simplified by directly heating the diazonium salt; this would also reduce the required amount of solvent. The important point here is the concentration of dil.H2SO4 during heating. A high concentration of dil.H2SO4 might lead to sulfonation as a side reaction,18) so we examined various concentrations up to 20% (Table 1, entries 1–4). In this experiment, the reaction mixture was diluted to about 50 µM, and the reaction progress was monitored by HPLC. While 15% dil.H2SO4 showed the highest area%, the yield was only 43% (Table 1, entry 3). Decreasing the concentration of substrate 3 did not lead to a significant improvement (Table 1, entry 5). As a side reaction of hydroxylation by means of the Sandmeyer reaction, dimer formation between the produced phenol and unreacted aniline has been reported.18) Heating the dil.H2SO4 after adding an organic solvent has been reported to prevent such dimerization and to increase the product yield.18) Since CPME works as the organic solvent in the Sandmeyer reaction,19) we examined CPME in the present reaction. When CPME was added to the dil.H2SO4 and the mixture was directly refluxed, the area% reached 72% (Table 1, entry 6). A small further improvement was obtained (73%) by decreasing the concentration of substrate 3, and finally we judged that the conditions of entry 7 were optimal for this reaction. On the other hand, the conditions of Entry 6 may also be noteworthy from the viewpoint of the amount of waste.
Table 1. Comparison of Conditions for
Steps a and
b| Entry | Conc. of H2SO4aq (v/v%) | Conc. of 3 (mol/L) | Solvent | Area% of 4a) |
|---|
| 1 | 5 | 0.25 | 5% H2SO4aq | 7% |
| 2 | 10 | 0.25 | 10% H2SO4aq | 27% |
| 3 | 15 | 0.25 | 15% H2SO4aq | 43% |
| 4 | 20 | 0.25 | 20% H2SO4aq | 18% |
| 5 | 15 | 0.125 | 15% H2SO4aq | 46% |
| 6 | 15 | 0.125 | 15% H2SO4aq/CPME | 72% |
| 7 | 15 | 0.084 | 15% H2SO4aq/CPME | 73% |
a) Area% was detected by HPLC.
Step c: O-alkylation
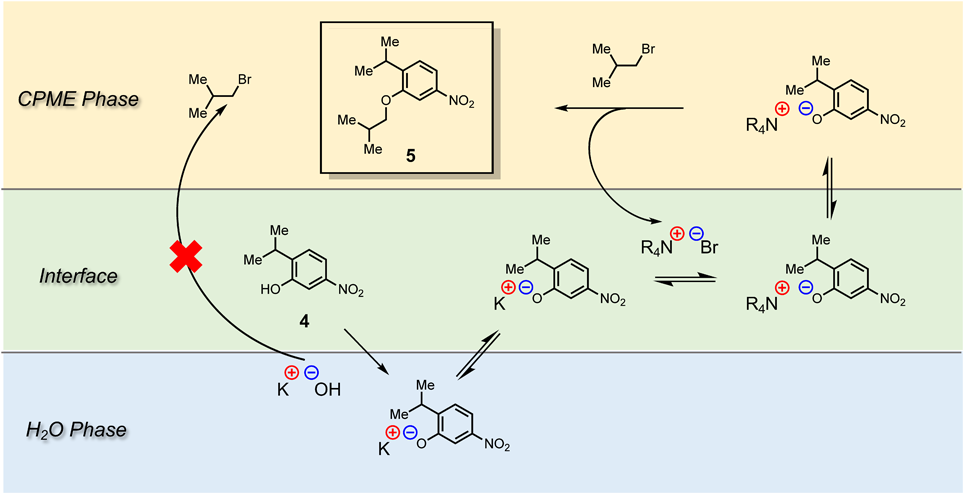
In the Medicinal Chemistry method, O-isobutoxylated substrate 5 was obtained in a yield of 71% by stirring for 2 h using dry DMF as a solvent, K2CO3 as a base, potassium iodide (KI) as a catalyst and isobutyl bromide as a reaction reagent.10) In this method, nitrophenol 4 was not completely eliminated, and purification by column chromatography was required.10) First, we tried CPME as a solvent, but the reaction did not reach completion (Table 2, entry 2). This reaction between phenoxide ion and isobutyl bromide is considered to proceed via an SN2 mechanism. NaOH, a stronger base than K2CO3, was used (Table 2, entry 3). However, under the conditions of entry 2, NaOH did not sufficiently dissolve in CPME, and the reaction did not proceed. Next, in order to dissolve NaOH, the same amount of H2O as CPME was added. Under this condition, KI was not added, because it is expected to dissolve in H2O. When the reaction was carried out at 70 °C for 24 h, a part of the starting material 4 still remained (Table 2, entry 4). The starting material 4 is considered to exist as a phenoxide ion in the H2O layer under basic conditions. On the other hand, isobutyl bromide is expected to exist in the CPME layer. Therefore, we focused on tetrabutylammonium bromide (TBAB), a phase-transfer catalyst that catalyzes the reaction at the interface between the organic layer and the H2O layer.20) Addition of a catalytic amount of TBAB significantly promoted the reaction and the reaction progress reached 56% after 24 h (Table 2, entry 5). As described above, CPME is known to azeotrope with H2O at 83 °C.17) The boiling point of isobutyl bromide is about 90 °C, and it was expected that reflux could be achieved by using CPME and H2O as solvents. When the reaction mixture obtained under the conditions of entry 5 was refluxed, the reaction progress reached 70% after 24 h (Table 2, entry 6). In order to completely eliminate the starting material 4, the amount of base was increased, and in this case the reaction was completed in 10 h (Table 2, entry 7). Since KOH is used as a base in Step f (N-alkylation), we also examined the use of KOH in this reaction (Table 2, entry 8). It is considered important to reduce the number of types of reagents used in process synthesis. The reaction was completed in 8 h under these conditions. The starting material 4 is considered to exist as a potassium salt in the H2O layer. The presence of the phase-transfer catalyst TBAB allows the potassium salt to become a quaternary amine salt, promoting the reaction with isobutyl bromide in the CPME layer. Furthermore, this was expected to prevent the loss of isobutyl bromide due to SN2 reaction with KOH21,22) (Fig. 2). Overall, the conditions of entry 8 were judged as optimal for Step c.
Table 2. Comparison of Conditions for
Step c| Entry | Base | Catalyst | Solvent | Internal temp. | Result a, b) |
|---|
| 1 | K2CO3 (1.2 equiv.) | KI (0.1 equiv.) | Dry DMF | 70 °C | Medicinal Chemistry method |
| 2 | K2CO3 (1.2 equiv.) | KI (0.1 equiv.) | CPME | 70 °C | No reaction |
| 3 | NaOH (1.2 equiv.) | KI (0.1 equiv.) | CPME | 70 °C | No reaction |
| 4 | NaOH (1.2 equiv.) | — | CPME/H2O | 70 °C | 17% progress at 24 h |
| 5 | NaOH (1.2 equiv.) | TBAB (0.1 equiv.) | CPME/H2O | 70 °C | 56% progress at 24 h |
| 6 | NaOH (1.2 equiv.) | TBAB (0.1 equiv.) | CPME/H2O | Reflux | 70% progress at 24 h |
| 7 | NaOH (2.4 equiv.) | TBAB (0.1 equiv.) | CPME/H2O | Reflux | Complete at 10 h |
| 8 | KOH (2.4 equiv.) | TBAB (0.1 equiv.) | CPME/H2O | Reflux | Complete at 8 h |
a) Area% was determined by HPLC. b) Reaction progress was confirmed every 2 h in all cases.
Step d: Reduction
In the Medicinal Chemistry method, Pd-catalytic reduction using acetic acid as a solvent was performed. Then, since the subsequent coupling reaction with 6-chloronicotininic acid is carried out under acidic conditions, the aniline 6 obtained by Pd-catalytic reduction was isolated as an acetate salt.10) Since Pd-catalytic reduction is potentially dangerous in process chemistry when the reaction is scaled up, this reduction was performed after sufficient degassing to remove oxygen.23) Reduction conditions using CPME as a solvent at normal pressure were examined, but without success. Therefore, in anticipation of promoting the reaction, we used EtOH, which is a protonic solvent. As a result, the starting material 5 disappeared and the target aniline 6 was obtained. Then, the solvent was changed from EtOH to CPME for the subsequent coupling reaction. In addition, p-toluenesulfonic acid (pTSA) salt 6b was selected because it was expected to be more soluble in CPME than the acetate salt 6a used in the Medicinal Chemistry method.
Step e: Coupling reaction
In the Medicinal Chemistry method, aminoacetate salt 6a was dissolved in acetic acid, and coupled with 6-chloronicotinic acid, then the solvent was replaced with MeOH and methyl ester 7a was obtained by esterification with H2SO4.10) In order to simplify this reaction, we aimed to obtain the ethyl ester 7b in one step by coupling amino pTSA salt 6b and ethyl 6-chloronicotinate in CPME. If H2O is present, hydrolysis may occur. It was confirmed that hydrolysis could be prevented by dehydration in a Dean–Stark apparatus before the reaction, and the ethyl ester 7b was obtained as a single product.
Step f and Step g: N-alkylation and hydrolysis
In the Medicinal Chemistry method, N-alkylation was performed by using EtI and NaH as a base, and the final compound 1 was synthesized by subsequent hydrolysis using MeOH and NaOHaq.10) In scaling up this reaction, NaH was considered unsuitable due to safety concerns, and different bases were examined. With CPME as a solvent, N-alkylation and hydrolysis of the ester might proceed simultaneously in the presence of H2O. Therefore, before the reaction, the CPME solution of ethyl ester 7b was sufficiently dehydrated using a Dean–Stark apparatus. As a base, KOH (used in Step c) was first examined (Table 3, entries 2, 3). To enable subsequent hydrolysis of the ester in one pot, a large excess of base was added. N-Alkylation proceeded at 60 °C, but not at room temperature. However, even at 60 °C, the reaction was not complete after 12 h, and hydrolysis continued over 24 h. This result suggested that extension of the reaction time may cause hydrolysis by H2O, which was thought to be produced by abstracting the proton of the secondary amine with a hydroxide ion. Therefore, we attempted to promote the N-alkylation rather than hydrolysis by adding TBAB (used in Step c). Under these conditions, the starting material 7b disappeared and the N-Et ethyl ester 8b was obtained in 6 h (Table 3, entry 4). When NaOH was employed, which was also used in Step c, hydrolysis proceeded at the same time (Table 3, entry 5). Since NaOH is highly deliquescent, it was considered that KOH, which is less deliquescent, would be more suitable as a base.24,25) At the interface between solid KOH and CPME, the reaction was promoted by TBAB, and the desired N-Et ethyl ester 8b was obtained21,22) (Fig. 3). Based on these results, the conditions of entry 4 were judged as the most suitable for Step f.
Table 3. Comparison of Conditions for
Step f| Entry | Base | Catalyst | Solvent | Internal temp. | Resulta, b) |
|---|
| 1 | NaH (1.2 equiv.) | — | DMF | r.t. | Medicinal Chemistry method |
| 2 | KOH (4.0 equiv.) | — | CPME | r.t. | Almost no reaction (approx.1%) |
| 3 | KOH (4.0 equiv.) | — | CPME | 60 °C | 30% progress at 12 h |
| Hydrolysis progressed over 24 h |
| 4 | KOH (4.0 equiv.) | TBAB (0.1 equiv.) | CPME | 60 °C | 100% progress at 6 h |
| 5 | NaOH (4.0 equiv.) | TBAB (0.1 equiv.) | CPME | 60 °C | 50% progress at 12 h |
| Hydrolysis progressed over 24 h |
a) Area% was determined by HPLC. b) Reaction progress was confirmed every 2 h in all cases. r.t.: room temperature.
In the final step of hydrolysis, CPME was evaporated under reduced pressure, and EtOH and H2O were added to a flask containing N-Et ethyl ester 8b and KOH. The mixture was refluxed to obtain the target compound 1, which was purified by recrystallization.
Synthesis on 100 mmol ScaleContinuous synthesis of 1 was performed using 100 mmol (13.5 g) of the starting material 2.
[Steps a and b] To a solution of 2 (13.5 g, 100 mmol) in conc.H2SO4 (180 mL), KNO3 (10.1 g, 100 mmol) was added to afford nitroaniline 3 with 99% purity (Supplementary Fig. S1). The obtained conc.H2SO4 solution of nitroaniline 3 was poured onto ice (1080 mL), and NaNO2 (7.25 g, 105 mmol) was added to the resulting solution on an ice bath to prepare the diazonium salt. CPME (400 mL) was added to the flask, and Sandmeyer reaction was initiated by heating. Since unreacted 3 and inorganic substances were present in the H2O layer, washing the CPME layer with H2O gave nitrophenol 4 with a purity of 65% (Supplementary Fig. S2).
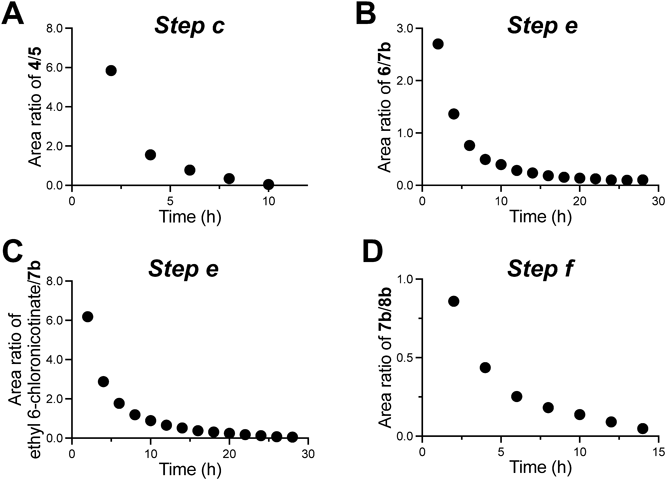
[Step c] Compound 4 obtained in the previous step was dissolved in CPME (100 mL) and H2O (100 mL). To the mixture were added KOH (13.5 g, 240 mmol), TBAB (3.22 g, 10 mmol) and isobutyl bromide (20.6 g, 150 mmol). After refluxing for 8 h the disappearance of nitrophenol 4 was confirmed (Fig. 4A). Since all reagents other than the desired product 5 were present in the H2O layer, the CPME layer was washed with H2O to afford 5. The combined aqueous layer afforded the main product with purity of 67% (Supplementary Fig. S3).
[Step d] After evaporation under reduced pressure, the solvent was replaced with EtOH (200 mL), and Pd-catalyzed reduction using H2 gas was performed. The disappearance of the starting material 5 was confirmed after reaction for 24 h. Then, EtOH was evaporated under reduced pressure and a CPME solution (100 mL) of aniline 6 was prepared. The concentration of aniline 6 was measured by HPLC, and an equimolar amount of pTSA was added. The mixture was stirred at room temperature to obtain 6b as pale brown needles with a purity of 85% (Supplementary Fig. S4).
[Step e] To the CPME solution of 6b was added ethyl 6-chloronicotinate (11.3 g, 60.7 mmol). The solution was refluxed for 21 h, during which time 6b disappeared (Fig. 4B, 4C). After the reaction, the CPME solution was washed with H2O to remove pTSA. The ethyl ester 7b was obtained as a black solid with a purity of 68% (Supplementary Figs. S5B, S5D). Residual ethyl 6-chloronicotinate was also observed. Recrystallization from EtOH gave 7b (17.5 g, 49.1 mmol) as a pale brown solid with a purity of 92% (Supplementary Fig. S5). It should be noted that it was difficult to achieve decolorization at the final step.
[Steps f and g] After dissolving ethyl ester 7b (17.5 g, 49.1 mmol) in CPME (200 mL), the solution was dehydrated in a Dean–Stark apparatus. Then, to the resulting solution were added KOH (11.0 g, 196 mmol), TBAB (1.58 g, 4.9 mmol) and EtI (9.17 g, 58.8 mmol) under an Ar atmosphere, and the mixture was heated at 60 °C for 14 h. Compound 7b disappeared and the N-Et ethyl ester 8b (Fig. 4D, Supplementary Fig. S6) was formed. CPME was removed under reduced pressure and EtOH and H2O (100 mL each) were added. The mixture was heated for 1 h to afford the final compound 1 as a pale brown solid, which was recrystallized from 70% EtOH to afford pale brown needles with a purity of 99%, in a total yield of 32% over 7 steps (Supplementary Fig. S7).
CPME was recovered and reused as much as possible during the 100 mmol-scale synthesis. At this scale, the amounts of waste CPME and EtOH were about 50 mL and 650 mL, respectively.
Measurement of the Amount of Waste and Calculation of the E-FactorWe were interested in evaluating how much more environmentally friendly the new process is, compared to the Medicinal Chemistry method. In multi-step synthesis, the E-factor is widely used as an index of environmental impact.26,27) This is defined with the weight (kg) of industrial waste (solvent, silica gel, etc.) produced to obtain 1 kg of the target product. In this calculation, water may or may not be included as waste. As there is a concern that the amount of waste would be increased excessively by including water, E-factors are calculated excluding waste water in many cases. For the Medicinal Chemistry method and the process method established above, we compared the effects of both methods on the environment by calculating the E-factor. In both methods, 20 mmol (2.7 g) of starting material 2 was used, and the amount of waste solvent in each step and the amount of silica gel (when column chromatography was carried out) were all calculated. The E-factor excluding water was 4669 for the Medicinal Chemistry method and 124 for the process method (Supplementary Figs. S8–S11, Table S2). Thus, the new process method showed an improvement of about 35 times compared to the Medicinal Chemistry method, and is therefore a more environmentally friendly synthetic method for 1. In general, the E-factor in pharmaceutical syntheses is between 25 and 200,26,27) so it can be said that the new process method is acceptable. When water was included in the waste, the E-factors of the two methods were 7559 and 390, respectively (Supplementary Table S3).
Process Method Using Other Hydrophobic Ether SolventsIn addition to CPME, 2-MeTHF and MTHP have been used as hydrophobic ether solvents for process chemistry. Thus, we evaluated whether the synthesis of 1 was possible using these solvents, starting from 20 mmol (2.7 g) of 2. Compound 1 was obtained in total yields of 7 and 50%, respectively (Supplementary Figs. S12, S13, Table S4). The low total yield for 2-MeTHF is considered to be due to the low yield in Step b (Sandmeyer reaction). The main product in Step b in 2-MeTHF was isolated and analyzed by 1H-NMR. The NMR chart revealed that 1-isopropyl-4-nitrobenzene (3′) was produced via the introduction of a proton into the diazonium site (Supplementary Fig. S14).28) The HPLC retention time of 3′ was consistent with that of the by-product in Step b of the 100 mmol scale synthesis using CPME. Although the safety of the solvent has not been evaluated yet,14) MTHP gave the highest yield and could be of interest. Nevertheless, CPME is inexpensive and easily available, and may be advantageous for use in process chemistry.
Conclusion
In this study, we aimed to establish a scaled-up process synthesis for RXR agonist NEt-3IB (1), a drug candidate for IBD, that would be consistent with SDGs. The only organic solvents used for reactions and extraction are a hydrophobic ether (CPME) and EtOH, and the ether can be azeotropically dehydrated and reused. Simple purification by recrystallization twice from EtOH afforded 99% pure 1 in a yield of over 30%. When CPME was used, the amounts of waste CPME and EtOH were about 50 and 650 mL, respectively, from a 100 mmol scale reaction, and the E-factor was at least 35 times lower than that of the Medicinal Chemistry method. This work provides a scaled-up green process for synthesizing 1, and we believe this work, which changes the solvent used to environmentally preferable ones based on the existing synthetic method for 1, also offers a useful illustration of how synthetic methods for small-molecular drugs can be adapted and improved to contribute to the SDGs.
Experimental
GeneralThe reagents used in this study were purchased from the indicated sources: 2-isopropylaniline (2) (Sigma-Aldrich, MO, U.S.A.), KNO3 (Wako, Osaka, Japan), conc.H2SO4 (Wako), NaNO2 (Kanto Kagaku, Tokyo, Japan), urea (TCI, Tokyo, Japan), CPME (Wako), 1-bromo-2-methylpropane (Sigma-Aldrich), KOH (Wako), NaOH (Sigma-Aldrich), TBAB (TCI), N2 gas (Chugoku Air Water, Japan), H2 gas (Chugoku Air Water), EtOH (Wako), p-toluenesulfonic acid monohydrate (TCI), ethyl 6-chloronicotinate (TCI), ethyl iodide (Wako), conc.HCl (Wako), 2-MeTHF (TCI), and MTHP (TCI).
The progress of each reaction was monitored by TLC on a 0.2 mm TLC plates (Merck, aluminium-backed, silica gel 60 F245) and spots were located under UV light. Silica gel 60 (particle size 0.035–0.070 mm) was used for flash column chromatography. The progress of each reaction was also monitored by HPLC. The HPLC system used was a Shimadzu liquid chromatographic system (Japan) consisting of a LC-20AT pump, SPD-20A detector, and CTO-10AS column oven. Data were processed using Labsolutions software. The reaction mixture (20 µL) was injected onto an Inertsil ODS-3 column (4.6 mm i.d. × 100 mm, 5 µm, GL Sciences, Tokyo, Japan) fitted with a guard column of Intersil ODS-3 (4.0 mm i.d. × 10 mm, 3 µm, GL Sciences) at 40 °C; the mobile phase was MeOH/H2O = 85/15 + 0.1% formic acid. The flow rate was 0.7 mL/min. A photodiode array (PDA) detector was used to monitor absorbance at 260 nm.
1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra were recorded on a Varian NMR System PS600 at room temperature. Deuterated chloroform (CDCl3) was used as the solvent for all routine NMR measurements. Chemical shifts are reported in ppm relative to the respective deuterated solvent peak, δ 7.26 ppm for 1H-NMR and δ 77.2 ppm for 13C-NMR, and coupling constants are given in Hz. FAB-MS spectra (low- and high-resolution mass spectra) were measured on a JEOL JMS-700 mass spectrometer.
SynthesisExamining the Conditions for Each StepExamining the Conditions of Step a and Step b (Table 1)Compound 2 was added to conc.H2SO4 (0.6 mL) in a 100 mL round-bottomed flask on ice and stirred to dissolve the solid. The solution was cooled below 5 °C, then KNO3 (1.0 mol equiv.) was added. The reaction mixture was stirred at 5 °C for 30 min, then poured onto ice and washed with H2O. NaNO2 (1.05 mol equiv.) was slowly added at below 5 °C. The presence of excess nitrite ion was checked by using potassium iodide starch paper. To the reaction mixture were added urea (0.125 mol equiv.) and CPME as indicated (+/−). The reaction mixture was refluxed for 1 h, then diluted to about 50 µM. The reaction progress was monitored by HPLC. The reaction conditions are listed in Supplementary Table S1.
Examining the Conditions of Step c (Table 2)To a solution of 3 (10 mmol) in solvent (CPME 20 mL or CPME/H2O = 10 mL/10 mL) in a 100 mL round-bottomed flask were added base (K2CO3 or NaOH or KOH, 12 mmol or 24 mmol), and catalyst (KI or TBAB, 1.0 mmol). The reaction mixture was heated at 60 °C for 30 min. Then 1-bromo-2-methylpropane (iBuBr, 15 mmol) was added, and the mixture was heated to 70 °C (internal temperature) or refluxed. The reaction mixture was diluted to about 50 µM and the reaction progress was monitored by HPLC.
Examining the Conditions of Step f (Table 3)To a solution of 7b (10 mmol) in CPME (20 mL) in a 100 mL round-bottomed flask were added base (NaOH or KOH, 40 mmol) and catalyst (TBAB 1.0 mmol or not), and the mixture was stirred at 60 °C for 30 min. Then EtI (12 mmol) was added and stirring was continued at 60 °C (internal temperature). The reaction mixture was diluted to about 50 µM and the reaction progress was monitored by HPLC.
100 mmol-Scale Synthesis[Step a] Conc.H2SO4 (180 mL) was poured to a 1-L 3-neck flask equipped with a mechanical stirrer and a thermometer, and cooled to below 5 °C. To the flask, 2-isopropylaniline (2, 13.5 g, 100 mmol) was added at 1 mL/min with stirring (250 rpm). After the addition, the precipitated solid was dissolved by increasing the temperature to r.t. After cooling the reaction mixture to below 5 °C again, powdered KNO3 (10.1 g, 100 mmol) was added slowly (1 g/min) with stirring (200 rpm). The progress of the reaction was monitored by HPLC (the total reaction time was 1 h). The retention times of 2 and 3 were 2.45 and 2.64 min, respectively (Supplementary Fig. S1).
[Step b] The reaction mixture was poured onto ice (300 mL) in a 500-mL beaker. The solution was transferred into a 1-L 3-neck flask and the beaker was washed with H2O (820 mL). The concentration of H2SO4aq at this stage was 15% (180 mL conc.H2SO4/1080 mL H2O). The solution was cooled to below 5 °C, and powdered NaNO2 (7.25 g, 105 mmol) was added slowly (200 mg/30 s) to the solution with stirring (200 rpm). The presence of excess nitrite ion was checked by the use of potassium iodide starch paper. To the reaction mixture were added urea (750 mg, 12.5 mmol) and CPME (400 mL with stirring (250 rpm) at 80 °C (internal temperature). After confirming completion of the reaction by HPLC (the total reaction time was 1 h), the reaction mixture was cooled to room temperature. The retention time of 4 was 3.25 min (Supplementary Fig. S2). The reaction mixture was poured into a separating funnel and the water layer was removed. The organic layer was washed with H2O (200 mL × 3) and evaporated under reduced pressure.
[Step c] To make the reaction solution basic (pH 11), H2O (100 mL) and KOH (13.5 g, 240 mmol) were added to 3 in a 1-L 3-neck flask. To the solution were added CPME (100 mL) and TBAB (3.22 g, 10 mmol), and the mixture was stirred (200 rpm) at 60 °C (internal temperature) for 2 h. Then, 1-bromo-2-methylpropane (20.6 g, 150 mmol) was added, and stirring was continued (200 rpm) at 80 °C (internal temperature). The progress of the reaction was monitored by HPLC (the total reaction time was 8 h). The retention time of 5 was 8.29 min (Supplementary Fig. S3). After the reaction was finished, the solution was cooled and then poured into a separating funnel. The organic layer was separated and washed with H2O (100 mL ×3). The CPME phase was evaporated under reduced pressure.
[Step d] After adding EtOH (200 mL) to 5 in a 1-L Erlenmeyer flask, the air in the flask was replaced with nitrogen gas. Then, Pd/C (1.19 g, 5% (w/w) of 5) was added to the flask, which was purged again with nitrogen. After replacement of N2 with H2 from a balloon, the solution was stirred at room temperature under an H2 atmosphere. The progress of the reaction was monitored by HPLC (the total reaction time was 24 h). The retention time of 6 was 1.81 min (Supplementary Fig. S4). After confirming completion of the reaction, the H2 atmosphere was replaced with nitrogen gas. The reaction mixture was filtered through Celite and washed with EtOH (50 mL). The filtrate was evaporated under reduced pressure to remove EtOH. The residue was dissolved in CPME (100 mL) and the concentration of the product was determined by HPLC (14.2 g, 68.6 mmol). Then, p-toluenesulfonic acid (13.0 g, 68.6 mmol) was added and the mixture was stirred for 1 h at room temperature, affording 6b as precipitated pale brown needles.
[Step e] A 1-L 3-neck flask was equipped with a mechanical stirrer, a thermometer, and a Dean–Stark apparatus with a Dimroth condenser. Before adding reagents, the CPME solution of 6b was heated to remove water using the Dean–Stark apparatus. The boiling temperature was initially 85 °C, rising to 105 °C (internal temperature) after the removal of water. Then, ethyl 6-chloronicotinate (12.7 g, 68.6 mmol) was added to the CPME solution of 6b and the reaction mixture was stirred (200 rpm) under reflux. The progress of the reaction was monitored by HPLC (the total reaction time was 28 h from the boiling). The retention time of 7b was 9.29 min (Supplementary Fig. S5). After confirming completion of the reaction by HPLC, the reaction mixture was poured into a separating funnel and washed with H2O (100 mL × 3). The CPME solution was concentrated under reduced pressure. The residue was recrystallized from EtOH (40 mL) and the precipitated solid was collected. Recrystallization was performed one more time from 70% EtOHaq (20 mL) and the precipitated solids were combined to yield 7b as a pale white solid (17.5 g, 49.1 mmol). 1H-NMR (400 MHz, CDCl3): δ = 8.81 (1H, dd, J = 2.3, 0.7 Hz), 8.05 (1H, dd, J = 8.8, 2.3 Hz), 7.31 (1H, s), 7.18 (1H, d, J = 7.8 Hz), 6.86–6.81 (3H, m), 4.35 (2H, q, J = 7.2 Hz), 3.71 (2H, d, J = 6.6 Hz), 3.33 (1H, seq, J = 7.4 Hz), 2.13 (1H, seq, J = 6.6 Hz), 1.38 (3H, t, J = 7.2 Hz), 1.23 (7H, d, J = 7.4 Hz), 1.06 (6H, d, J = 6.6 Hz). 13C-NMR (150 MHz, CDCl3): δ = 165.73, 159.16, 157.27, 150.93, 139.32, 137.24, 134.15, 126.82, 117.18, 114.32, 106.79, 106.13, 74.52, 60.91, 28.66, 26.98, 22.82, 19.61, 14.59. FAB-MS m/z: 357 [M + H]+. HRMS (FAB+) m/z: [M + H]+ Calcd for C21H28N2O3, 357.2657. Found, 357.2128.
[Step f] Solid 7b from the previous step was dissolved in CPME (200 mL) and the solution was added to a 1-L 3-neck flask equipped with a mechanical stirrer, a thermometer, and a Dean–Stark apparatus with a Dimroth condenser. After removal of water by the Dean–Stark apparatus, KOH (11.0 g, 196.0 mmol) and TBAB (1.58 g, 4.9 mmol) were added. The mixture was stirred (200 rpm) at 50 °C (internal temperature) under an Ar atmosphere for 1 h, then EtI (9.17 g, 58.8 mmol) was added, and stirring was continued at 60 °C (internal temperature) under an Ar atmosphere. The progress of this reaction was monitored by HPLC (the total reaction time was 14 h). The retention time of 8b was 19.72 min (Supplementary Fig. S6). The reaction mixture was evaporated under reduced pressure.
[Step g] EtOH (50 mL) and H2O (50 mL) were added to 8a in a 1-L 3-neck flask equipped with a mechanical stirrer, a thermometer and a Dimroth condenser, and the mixture was stirred (200 rpm) at 80 °C (internal temperature). The progress of reaction was monitored by HPLC (the total reaction time was 1 h). The retention time of 1 was 9.40 min (Supplementary Fig. S7). The reaction mixture was transferred to a 500-mL eggplant flask, and the EtOH was evaporated under reduced pressure. To acidify the solution to pH 4, conc.HCl (10 mL) was added and precipitated solid was filtered with the aid of water (50 mL) using a suction funnel. The solid was dried and recrystallized overnight from 70% EtOHaq (750 mL) with stirring at r.t. The collected crystals were dried overnight under reduced pressure at 40 °C to give 11.5 g of NEt-3IB (1) as pale brown needles (32.3 mmol, 99% purity by HPLC). 1H-NMR (600 MHz, CDCl3): δ = 8.93 (1H, d, J = 2.2 Hz), 7.84 (1H, dd, J = 9.1, 2.2 Hz), 7.26 (1H, d, J = 8.0 Hz), 6.77 (1H, dd, J = 8.0, 2.0 Hz), 6.64 (1H, d, J = 2.0 Hz), 6.25 (1H, d, J = 9.1 Hz), 4.05 (2H, q, J = 7.0 Hz), 3.68 (2H, d, J = 6.5 Hz), 3.36 (1H, seq, J = 6.5 Hz), 2.12 (1H, seq, J = 6.7 Hz), 1.26 (6H, d, J = 6.5 Hz), 1.25 (3H, t, J = 7.0 Hz), 1.06 (6H, d, J = 6.7 Hz). 13C-NMR (150 MHz, CDCl3): δ = 171.59, 160.87, 157.54, 152.10, 141.85, 137.75, 136.30, 127.17, 119.61, 113.37, 110.60, 107.58, 74.34, 45.50, 28.45, 26.87, 22.55, 19.38, 13.09. mp 189.6–191.0 °C. FAB-MS m/z: 357 [M + H]+. HRMS (FAB+) m/z: [M + H]+ Calcd for C21H28N2O3, 357.2666. Found, 357.2178.
Anal. Calcd for C21H28N2O3: C, 70.76; H, 7.92; N, 7.86. Found: C, 71.02; H, 8.09; N, 7.88.
20 mmol-Scale SynthesisCompound 2 (2.70 g, 20 mmol) was used as a starting material.
Medicinal Chemistry MethodAll synthetic steps were performed according to reference S1. The final compound 1 was obtained in a yield of 0.91 g (2.56 mmol) (13%).
Process Method (using CPME)All synthesis were performed as described above. The final compound 1 was obtained in a yield of 1.95 g (5.46 mmol) (27%).
Comparison of E-Factors for the Medicinal Chemistry and Process MethodsAll the weights of waste materials, including waste solvent, either including or excluding water, were calculated. The E-factors were evaluated according to the following equation.
Process Method (using 2-MeTHF)All synthetic procedures were performed as described above, except that 2-MeTHF was used instead of CPME. The final compound 1 was obtained in a yield of 0.51 g (1.43 mmol) (7%). mp 191.5–192.7 °C.
Process Method (using MTHP)All synthetic procedures were performed as described above, except that MTHP was used instead of CPME. The final compound 1 was obtained in a yield of 3.56 g (9.98 mmol) (50%). mp 180.1–181.3 °C.
Acknowledgments
The authors are grateful to the Division of Instrumental Analysis, Okayama University for the NMR and MS measurements.
This work was partially supported by Grants from Okayama University Science, Technology, and Innovation Creation Fellowship (to Y.T.).
Author Contributions
Y.T., K.M. and H.K. conceived and designed the project. Y.T., S.K., and M.W. examined each reaction condition. Y.T. and K.M. performed 100 mmol scale synthesis. Y.T. performed 20 mmol scale synthesis and calculated E-factors. The manuscript was written by Y.T. and H.K.
Conflict of Interest
This study received funding from AIBIOS K.K. The founder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests.
Supplementary Materials
This article contains supplementary materials.
Notes
This paper is dedicated to Dr. Koichi Shudo, who passed away on 7 July 2021.
References
- 1) “The Sustainable Development Agenda.”: ‹https://www.un.org/sustainabledevelopment/development-agenda/›, cited 8 February, 2021.
- 2) Nabukalu J. B., Asamani J. A., Nabyonga-Orem J., Int. J. Health Policy Manag., 9, 297–308 (2020).
- 3) Seyedian S. S., Nokhostin F., Malamir M. D., J. Med. Life, 12, 113–122 (2019).
- 4) Janakiram N. B., Mohammed A., Qian L., Choi C. I., Steele V. E., Rao C. V., Neoplasia, 14, 159–168 (2012).
- 5) Mukherjee R., Davies P. J. A., Crombie D. L., Bischoff E. D., Cesario R. M., Jow L., Hamann L. G., Boehm M. F., Mondon C. E., Nadzan A. M., Paterniti J. R. Jr., Heyman R. A., Nature (London), 386, 407–410 (1997).
- 6) Mariani M. M., Malm T., Lamb R., Jay T. R., Neilson L., Casali B., Medarametla L., Landreth G. E., Sci. Rep., 7, 42270 (2017).
- 7) McFarland K., Spalding T. A., Hubbard D., Ma J. N., Olsson R., Burstein E. S., ACS Chem. Neurosci., 4, 1430–1438 (2013).
- 8) Sherman S. I., Clin. Lymphoma, 3, 249–252 (2003).
- 9) Standeven A. M., Escobar M., Beard R. L., Yuan Y. D., Chandraratna R. A., Biochem. Pharmacol., 54, 517–524 (1997).
- 10) Takamatsu K., Takano A., Yakushiji N., Morohashi K., Morishita K., Matsuura N., Makishima M., Tai A., Sasaki K., Kakuta H., ChemMedChem, 3, 780–787 (2008).
- 11) Kakuta H., Ohsawa F., Yamada S., Makishima M., Tai A., Yasui H., Yoshikawa Y., Biol. Pharm. Bull., 35, 629–633 (2012).
- 12) Kobayashi T., Furusawa Y., Yamada S., Akehi M., Takenaka F., Sasaki T., Akahoshi A., Hanada T., Matsuura E., Hirano H., Tai A., Kakuta H., ACS Med. Chem. Lett., 6, 334–338 (2015).
- 13) Matsumoto R., Takahashi D., Watanabe M., Nakatani S., Takamura Y., Kurosaki Y., Kakuta H., Hase K., Front. Pharmacol., 12, 715752 (2021).
- 14) Prat D., Pardigon O., Flemming H. W., Letestu S., Ducandas V., Isnard P., Guntrum E., Senac T., Ruisseau S., Cruciani P., Hosek P., Org. Process Res. Dev., 17, 1517–1525 (2013).
- 15) Antonucci V., Coleman J., Ferry J. B., Johnson N., Mathe M., Scott J. P., Xu J., Org. Process Res. Dev., 15, 939–941 (2011).
- 16) Kobayashi S., Tamura T., Yoshimoto S., Kawakami T., Masuyama A., Chem. Asian J., 14, 3921–3937 (2019).
- 17) Azzena U., Carraro M., Pisano L., Monticelli S., Bartolotta R., Pace V., ChemSusChem, 12, 40–70 (2019).
- 18) Yu Z., Ye X., Xu Q., Xie X., Dong H., Su W., Org. Process Res. Dev., 21, 1644–1652 (2017).
- 19) Taniguchi T., Imoto M., Takeda M., Nakai T., Mihara M., Iwai T., Ito T., Mizuno T., Nomoto A., Ogawa A., Heteroatom Chem., 26, 411–416 (2015).
- 20) Johnson C. R., Ansari M. I., Coop A., ACS Omega, 3, 10886–10890 (2018).
- 21) Makosza M., Ludwikow M., Rocz. Chem., 39, 1223 (1965).
- 22) Kitamura M., Shirakawa S., Maruoka K., Angew. Chem. Int. Ed., 44, 1549–1551 (2005).
- 23) Gallagher W. P., Marlatt M., Livingston R., Kiau S., Muslehiddinoglu J., Org. Process Res. Dev., 16, 1665–1668 (2012).
- 24) Scapino L., Zondag H. A., Bael J. V., Diriken J., Rindt C. C. M., Renew. Sustain. Energy Rev., 76, 1314–1331 (2017).
- 25) Konings R. J. M., Cordfunke E. H. P., J. Chem. Thermodyn., 20, 103–108 (1988).
- 26) Tobiszewski M., Marć M., Gałuszka A., Namieśnik J., Molecules, 20, 10928–10946 (2015).
- 27) Sheldon R. A., Green Chem., 9, 1273–1283 (2007).
- 28) Sharma Y., Nikam A. V., Kulkarni A. A., Org. Process Res. Dev., 23, 170–176 (2019).