Experimental
General Experimental ProceduresAll reactions were monitored by TLC using Merck 60 F254 precoated silica gel plates (0.25 mm thickness). UV spectra were recorded in MeOH on a JASCO V-560 instrument. Specific optical rotations were measured using a JASCO P-2200 and P-1020 polarimeter. Circular dichroism (CD) spectra were recorded on a JASCO J-1100 spectrometer. Fourier transform (FT)IR spectra were recorded on a JASCO FT/IR-4700 and SHIMADZU IR Affinity-IS. 1H- and 13C-NMR spectra were recorded on an ECX 500 (500 MHz for 1H-NMR, 125 MHz for 13C-NMR) and an ECZ 600 (600 MHz for 1H-NMR, 150 MHz for 13C-NMR) FT-NMR spectrometer. Data for 1H-NMR are reported as chemical shifts (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, dd = double doublet, ddd = double double doublet, m = multiplet, br = broad), coupling constant (Hz), integration, and assignment. Data for 13C-NMR are reported as chemical shifts. The HR mass spectra were recorded on a JEOL AccuTOF LC-plus JMS-T100LP and BRUKER impact II. HPLC was performed on a Shimadzu LC-20AT system coupled with an SPD M20A photodiode array detector (Shimadzu Co., Kyoto, Japan), using a CAPCELLPAK C18 (5 µm, 250 × 20.0 mm i.d., SHISEIDO Inc., Tokyo, Japan). Recycle HPLC was performed on a Japan Analytical Industry LC-9225 NEXT SERIES system coupled with a UV-600 NEXT detector and RI-700 NEXT detector (Japan Analytical Industry Co., Tokyo, Japan), using an Asahipak GS-510 20G and Asahipak GS-310 20G column (500 × 20.0 mm i.d., Shodex Inc., Tokyo, Japan). Preparative thin layer chromatography (PTLC) was performed using Merck 60 F254 precoated silica gel plates (0.25 mm thickness).
Plant MaterialThe heartwood of Adina rubescens was collected in Bogor Botanical Garden, Indonesia, in February 2017.
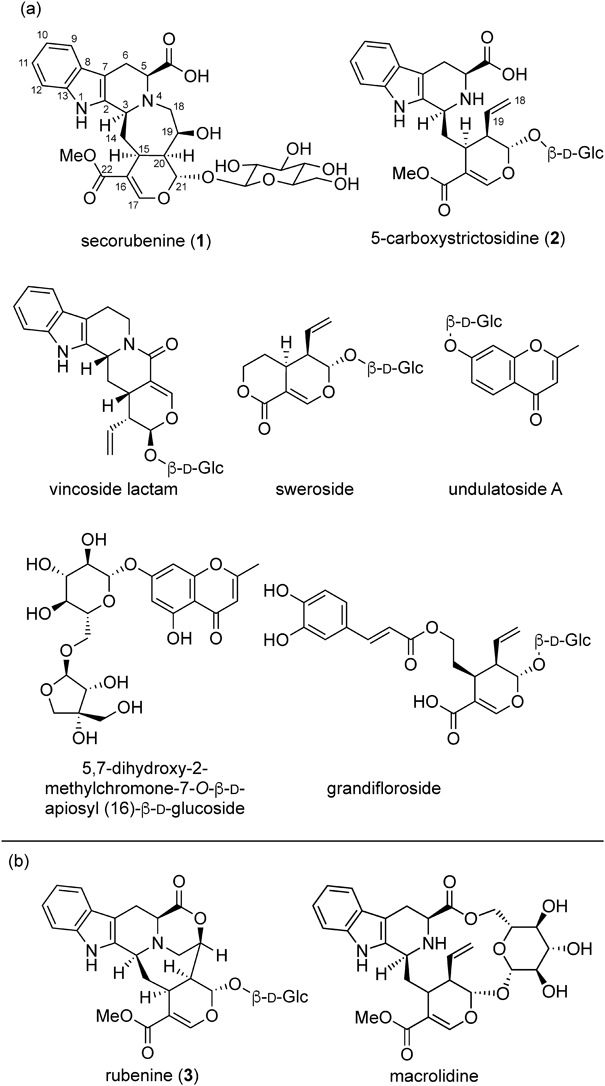
Extraction and IsolationThe powdered heartwood (1.6 kg dry weight) of Adina rubescens was macerated with MeOH (three times at room temperature for 24 h, total 12 L) to give the extract (40 g). A portion of the MeOH extract (5.3 g) was dissolved in water (1.6 L) and extracted with n-hexane (1.6 L ×3), with AcOEt (1.6 L ×3), and then with n-BuOH (1.6 L ×3) to give the n-hexane extract (49.5 mg), AcOEt extract (901.8 mg), and n-BuOH extract (2.288 g), respectively. The n-BuOH extract (2.288 g) was separated by HPLC (column: SHISEIDO CAPCELL PAK C18) with a MeCN/H2O gradient (20–60%) to give 15 fractions (fr): fr 1 (193.1 mg), fr 2 (38.8 mg), fr 3 (45.4 mg), fr 4 (22.6 mg), fr 5 (72.1 mg), fr 6 (60.4 mg), fr 7 (25.1 mg), fr 8 (31.3 mg), fr 9 (51.5 mg), fr 10 (78.1 mg), fr 11 (35.2 mg), fr 12 (61.0 mg), fr 13 (191.3 mg), fr 14 (36.6 mg), and fr 15 (87.1 mg). Fraction 11 (35.2 mg) was purified by recycling SEC-HPLC (column: Asahipak GS-510 20G and GS-310 20G) with MeOH to afford secorubenine (1, 8.9 mg). The known compounds isolated from the n-BuOH extract were 5-carboxystrictosidine (5.3 mg), vincoside lactam (0.6 mg),17) sweroside (5.6 mg),18) grandifloroside (27.5 mg),19) undulatoside A (2.0 mg),20) and 5,7-dihydroxy-2-methylchromone-7-O-β-D-apiosyl (16)-β-D-glucoside (15.3 mg).21)
Secorubenine (1): Pale yellow amorphous powder; [α]D23 –62 (c 0.34, MeOH); UV (MeOH) λmax nm 290.5, 281.5, 223.5; IR (attenuated total reflectance (ATR)) cm−1 3282, 2924, 1627, 1259; 1H-NMR (500 MHz, (CD3)2SO) δ: 7.37 (1H, s, H-17), 7.36 (1H, d, J = 7.5 Hz, H-9), 7.29 (1H, d, J = 7.5 Hz, H-12), 7.01 (1H, dd, J = 7.5, 7.5 Hz, H-11), 6.94 (1H, dd, J = 7.5, 7.5 Hz, H-10), 5.69 (1H, br d, J = 4.5 Hz, H-21), 4.53 (1H, d, J = 10.0 Hz, H-1′), 4.20 (1H, m, H-19), 4.15 (1H, m, H-3), 3.68 (3H, s, CO2Me), 3.59 (1H, overlapped, H-5), 3.59 (1H, overlapped, H-6′), 3.45 (1H, m, H-6′), 3.18–3.15 (2H, overlapped, H-3′, 5′), 3.08–2.90 (3H, overlapped, H-15, 2′, 4′), 2.90 (1H, m, H-18), 2.81 (3H, overlapped, H2-6, H-18), 2.55 (2H, overlapped, H-14, 20), 1.75 (1H, ddd, J = 12.0, 12.0, 12.0 Hz, H-14); 13C-NMR (125 MHz, (CD3)2SO) δ: 174.8 (CO2H), 167.1 (CO2Me), 151.8 (C-17), 136.3 (C-13), 134.9 (C-2), 126.6 (C-8), 120.9 (C-11), 118.7 (C-10), 117.8 (C-9), 111.7 (C-16), 111.4 (C-12), 106.4 (C-7), 98.8 (C-1′), 96.9 (C-21), 77.4 (C-5′), 76.6 (C-3′), 73.1 (C-2′), 70.0 (C-4′), 69.5 (C-19), 63.9 (C-5), 61.0 (C-6′), 58.4 (C-3), 53.0 (C-18), 51.4 (CO2Me), 41.9 (C-20), 34.4 (C-14), 29.9 (C-15), 22.7 (C-6); 1H-NMR (600 MHz, CD3OD, at 60 °C) δ: 7.56 (1H, s, H-17), 7.43 (1H, d, J = 7.5 Hz, H-9), 7.35 (1H, d, J = 7.5 Hz, H-12), 7.11 (1H, dd, J = 7.5, 7.5 Hz, H-11), 7.03 (1H, dd, J = 7.5, 7.5 Hz, H-10), 5.64 (1H, d, J = 8.4 Hz, H-21), 4.80 (1H, d, J = 8.4 Hz, H-1′), 4.62 (1H, overlapped, H-19), 4.59 (1H, overlapped, H-3), 4.00 (1H, m, H-5), 3.99 (1H, d, J = 12.0 Hz, H-6′), 3.86 (1H, overlapped, H-18), 3.79 (3H, s, CO2Me), 3.68 (1H, br dd, J = 12.0, 4.2 Hz, H-6′), 3.47 (1H, m, H-18), 3.36–3.25 (6H, overlapped, H2-6, H-2′, 3′, 4′, 5′), 3.24 (1H, m, H-15), 2.68 (1H, d, J = 15.0 Hz, H-14), 2.30 (1H, overlapped, H-20), 2.26 (1H, overlapped, H-14); 13C-NMR (150 MHz, CD3OD, at 60 °C) δ: 175.1 (CO2H), 169.1 (CO2Me), 153.9 (C-17), 138.5 (C-13), 131.5 (C-2), 127.6 (C-8), 123.0 (C-11), 120.4 (C-10), 119.0 (C-9), 112.4 (C-12), 111.1 (C-16), 107.7 (C-7), 101.9 (C-1′), 98.3 (C-21), 78.4 (C-5′), 78.0 (C-3′), 74.7 (C-2′), 71.4 (C-4′), 69.5 (C-5), 66.2 (C-3), 64.5 (C-19), 62.4 (C-6′), 57.0 (C-18), 52.0 (CO2Me), 44.3 (C-20), 34.9 (C-14), 33.8 (C-15), 25.9 (C-6); HRMS (ESI): found: 591.2172 [M + H]+, calcd. for C28H35N2O12: 591.2190.
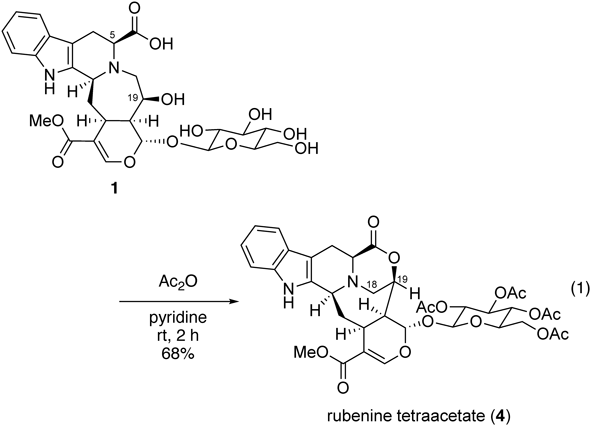
Chemical Modification and Total SynthesisAcetylation of Secorubenine (1)To a solution of secorubenine (1, 2.7 mg, 0.00457 mmol) in pyridine (30 µL), an excess amount of acetic anhydride (20 µL) was added at r.t. under Ar atmosphere. The reaction mixture was stirred for 2 h at r.t. The resulting mixture was concentrated under reduced pressure. The crude materials were purified by PTLC (70% AcOEt/n-hexane) to afford rubenine tetraacetate (4, 2.3 mg, 68%) as a pale yellow amorphous powder. All spectral data were identified based on our previously synthesized 4.
Rubenine tetraacetate (4): Pale yellow amorphous powder; [α]25D –30.0 (c 0.12, CHCl3); IR (neat) νmax cm−1 1745, 1217, 1036, 744; HRMS (ESI) [M + H]+ Calcd. for [C36H41N2O15]+: 741.2507, Found: 741.2529; 1H-NMR (600 MHz, CDCl3) δ: 7.48 (1H, d, J = 6.0 Hz), 7.44 (1H, s), 7.27 (1H, d, J = 6.0 Hz), 7.13 (1H, dd, J = 6.0, 6.0 Hz), 7.07 (1H, dd, J = 6.0, 6.0 Hz), 5.34 (1H, d, J = 12.0 Hz), 5.24 (2H, overlapped), 5.14 (1H, dd, J = 12.0, 12.0 Hz), 5.08 (1H, dd, J = 12.0, 12.0 Hz), 4.98 (1H, d, J = 6.0 Hz), 4.42 (1H, d, J = 6.0 Hz), 4.28 (2H, m), 4.19 (1H, d, J = 6.0 Hz), 3.96 (1H, d, J = 6.0 Hz), 3.88 (1H, dd, J = 12.0, 6.0 Hz), 3.78 (3H, s), 3.77 (1H, overlapped), 3.64 (1H, d, J = 18.0 Hz), 3.48 (1H, d, J = 18.0 Hz), 3.24 (1H, dd, J = 9.0, 9.0 Hz), 3.04 (1H, dd, J = 12.0, 6.0 Hz), 2.07 (1H, overlapped), 2.051 (3H, s), 2.047 (3H, s), 2.011 (3H, s), 2.007 (3H, s), 1.92 (1H, m); 13C-NMR (150 MHz, CDCl3) δ: 170.9, 170.3, 169.8, 169.60, 169.59, 167.5, 152.1, 136.3, 133.2, 126.8, 122.1, 119.5, 118.7, 110.8, 109.2, 105.2, 99.1, 96.7, 72.8, 72.3, 71.2, 68.5, 62.0, 58.9, 55.8, 51.7, 50.1, 43.9, 37.9, 32.5, 29.8, 20.9, 20.8, 20.73, 20.70, 20.2.
Preparation of Compound 8To a solution of L-tryptophan methyl ester (6, 25.3 mg, 0.116 mmol) and secologanin derivative 515) (33.2 mg, 0.0580 mmol) in CH2Cl2 (966 µL), powdered MS 4 Å (66.4 mg) was added at r.t. under Ar atmosphere. The resulting mixture was stirred for 1 h at r.t., followed by the addition of trifluoroacetic acid (22.2 µL, 0.290 mmol) at −20 °C. The reaction mixture was allowed to warm to 0 °C and then stirred for 1 h at this temperature. The resulting mixture was quenched with Et3N (81.0 µL) at 0 °C and filtered through a Celite pad, with CHCl3. The filtrate was concentrated under reduced pressure. The Pictet–Spengler reaction afforded the desired 3S-isomer 7, predominantly in quantitative conversion (C3S:C3R = 2.5 : 1), as determined by the crude NMR. The crude materials of 7 were directly loaded on PTLC (Wakogel® B-5F), where any remaining solvent was removed by drying with a blower. After 6 h, the silica gel was eluted with 10% MeOH/CHCl3. The resulting mixture was concentrated under reduced pressure and the resulting residue was purified by PTLC (SiO2, 60% AcOEt/n-hexane) to afford the desired product 8 (27.1 mg, 60% yield over two steps).
Compound 8: Pale yellow amorphous powder; [α]24D –44.3 (c 0.25, CHCl3); IR (neat) νmax cm−1 2922, 1744, 1705, 1638, 1435, 1368, 1219, 1034, 772, 743; HRMS (ESI) [M + H]+ Calcd. for [C37H45N2O16]+: 773.2764, Found: 773.2728; 1H-NMR (500 MHz, benzene-d6) δ: 7.53 (1H, s, H-17), 7.48–7.03 (4H, m, H-9, 10, 11, 12), 6.23 (1H, d, J = 8.5 Hz, H-21), 5.52 (1H, dd, J = 9.5, 9.5 Hz, H-3′), 5.41 (1H, dd, J = 9.5, 8.0 Hz, H-2′), 5.31 (1H, dd, J = 9.5, 9.5 Hz, H-4′), 5.13 (1H, d, J = 8.0 Hz, H-1′), 4.31 (1H, dd, J = 12.0, 5.0 Hz, H-6′), 4.28 (1H, br s, H-19), 4.08 (1H, dd, J = 12.0, 2.5 Hz, H-6′), 3.58 (1H, d, J = 10.5 Hz, H-3), 3.53–3.50 (1H, m, H-5′), 3.46 (3H, s, 22-CO2Me), 3.44 (1H, dd, J = 10.0, 4.5 Hz, H-5), 3.30 (3H, s, 5-CO2Me), 3.01–2.93 (3H, m, H-6, 15 and 18), 2.86 (1H, dd, J = 15.0, 3.5 Hz, H-6), 2.69 (1H, dd, J = 12.5, 4.5 Hz, H-18), 2.40 (1H, d, J = 11.0 Hz, H-14), 2.33–2.30 (1H, m, H-20), 1.95–1.87 (1H, m, H-14), 1.80 (3H, s, 6′-OCOMe), 1.73 (3H, s, 4′-OCOMe), 1.71 (3H, s, 3′-OCOMe), 1.66 (3H, s, 2′-OCOMe); 13C-NMR (125 MHz, benzene-d6) δ: 173.6 (CO2Me), 170.1 (6′-OCOMe), 169.9 (4′-OCOMe), 169.1 (3′-OCOMe), 169.1 (2′-OCOMe), 167.2 (C-22), 152.1 (C-17), 136.8 (C-8), 134.1 (C-2), 127.2 (C-13), 122.1 (C-11), 120.0 (C-10), 118.4 (C-9), 112.0 (C-12), 111.4 (C-16), 106.9 (C-7), 99.3 (C-21), 98.9 (C-1′), 73.1 (C-2′), 72.6 (C-5′), 71.8 (C-3′), 69.0 (C-19), 68.7 (C-4′), 63.9 (C-5), 61.8 (C-6′), 58.2 (C-3), 56.6 (C-18), 51.6 (C-24), 51.0 (C-25), 45.2 (C-20), 39.2 (C-14), 33.2 (C-15), 25.2 (C-6), 20.4 (6′-OCOMe), 20.3 (4′-OCOMe), 20.2 (3′-OCOMe), 20.2 (2′-OCOMe).
To a solution of 8 (27.0 mg, 0.0349 mmol) in MeOH (700 µL), 1M aqueous NaOH solution (700 µL) was added at 0 °C under Ar atmosphere. The reaction mixture was stirred for 2 h at 0 °C. The resulting mixture was quenched with 1M aqueous HCl solution (700 µL) and concentrated by lyophilization under reduced pressure. PTLC (SiO2, 30% MeOH/CHCl3) followed by HPLC (CAPCELLPAK C18, 1.0 mL/min, 20–70% MeCN/H2O gradient) afforded (−)-secorubenine (1, 15.2 mg, 74%) as a pale yellow amorphous powder; [α]D24 –91 (c 0.049, MeOH); 1H- and 13C-NMR spectra were compared as HCl salt of 1; see details in supporting information.