Current Topics: Reviews
Development of an Antigen Delivery System for a B Cell-Targeted Vaccine as an Alternative to Dendritic Cell-Targeted Vaccines
2022 年 70 巻 5 号 p. 341-350
詳細
2022 年 70 巻 5 号 p. 341-350
Vaccines have contributed to the prevention of infectious diseases for a long time. Pathogen-derived antigens and adjuvants in vaccine formulations stimulate immune cells to elicit humoral and cellular immune responses against pathogens. Achieving highly immune responses with decreased adverse effects requires the development of a system that can deliver antigens to specific immune cells. Dendritic cells (DCs) are well-known professional antigen presenting cells (APCs) that initiate acquired immune responses by presenting antigens to T cells. Accordingly, DC-targeted vaccines have been investigated and applied in clinical trials for the treatment of infectious diseases and for chronic diseases such as cancers. In addition to DCs, B lymphocytes are regarded as professional APCs despite their primary role in humoral immunity. Therefore, B cell-targeted vaccines are also expected to elicit both humoral and cellular immune responses. In this review we summarize the basic functions of DCs and B cells as APCs. We also provide information on DC and B cell targeted vaccines in preclinical and clinical settings. Finally, we introduce our novel antigen delivery system that targets splenic marginal zone B cells and the ability of this system to act as a novel vaccine that elicits both humoral and cellular immune responses.
Vaccines have made a significant contribution to public health since Edward Jenner first developed a smallpox vaccine in the 18th century. Thereafter, a variety of vaccines have been developed for the prevention of infectious diseases such as polio, measles, and pneumonia.1) The latest example is a vaccine against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that emerged in December 2019 as a novel coronavirus that caused a global pandemic—coronavirus disease 2019 (COVID-19).2) The emergence of SARS-CoV-2 has severely affected social and economic activities, and, as of this writing (early 2022), the spread of SARS-CoV-2 has yet to be completely controlled. However, mRNA-based vaccines against SARS-CoV-2 were created within only one year after the emergence of SARS-CoV-2.3,4) The vaccines are effective at decreasing onset and disease activity and are expected to suppress the pandemic. In addition, vaccines are applied not only to prevent infectious diseases but also to treat chronic diseases such as hypertension, diabetes, and cancers.5,6) Therefore, the demand for new vaccine development is continuously increasing.
Vaccines are composed of antigens, adjuvants and delivery systems. Antigens derived from pathogenic bacteria and viruses stimulate B cells and T cells to produce antigen-specific acquired immune responses (1st signal). In the absence of optimal stimulation, immune cells get trapped in immune tolerance, so adjuvants simultaneously provide danger signals via the activation of innate immunity (2nd signal).7) Delivery systems enable antigens and adjuvants to target immune cells, which maximizes efficacy and minimizes the systemic adverse effects of vaccines.8) Acquired immune responses are divided into B cell-mediated humoral responses and T cell-mediated cellular responses. B cells directly recognize antigens through B cell receptors (BCRs) and produce antibodies.9) On the other hand, T cells recognize antigen-derived peptides presented by antigen presenting cells (APCs) such as dendritic cells (DCs), macrophages, and B cells.10) APC-mediated activation of T cells also contributes to antibody production. Accordingly, it is essential to deliver antigens to APCs in order to maximize the humoral and/or cellular immune responses in vaccines.
Among several types of APCs, DCs are the most famous and promising version due to their high capacity for antigen presentation and high expression of co-stimulatory molecules such as CD80/CD86. In addition, a certain subset of DCs possess a unique property, which is referred to as cross-presentation.11) Exogenous antigens are generally presented onto major histocompatibility complex (MHC) class 2 molecules to activate helper T cells rather than killer T cells. However, certain DCs are able to cross-present exogenous antigens onto MHC class 1 molecules, which activates killer T cells. The ability to induce both responses makes DCs ideal target cells for vaccines.12) On the other hand, B cell-targeted vaccines have recently attracted interest as alternatives to DC-targeted versions.13) In addition to the original function of antibody production, B cells are known to serve as APCs that contribute to T cell-mediated cellular immunity.14,15)
This review first briefly summarizes the basic characteristics of DCs and B cells. Then, we present an overview of the trials of DC- and B cell-targeted vaccines, which includes cell-based vaccines. Finally, we introduce the concept of our novel splenic B cell-targeted vaccines and describe the effect in animals.
DCs, so-called professional APCs, play a pivotal role in the initiation of immune responses. DCs are functionally classified into two groups, plasmacytoid DCs (pDCs) and conventional DCs (cDCs).16) The former immediately produces a massive type 1 interferon (IFN) response to viral infection via the activation of toll-like receptors (TLRs). The latter is further divided into conventional DC type 1 (cDC1; CD11c+, CLEC9A+, XCR1+, CD8+) and type 2 (cDC2; CD11c+, CD11b+, SIRPα+, CD4+). cDC1 is a primary subset for the induction of CD8+ T cells by utilizing the function of cross-presentation, while cDC2 can prime CD4+ T cells that help antibody production from B cells. The function of each DC is well-correlated with their localization in the lymph nodes and spleen17) (Fig. 1); cDC1 is located in a deep T cell zone where CD8+ T cells exist, and cDC2 is located at the T-B border where CD4+ T cells exist. Such segregation of DCs promotes efficient T cell priming and determines the polarization of immune responses.

Antigen uptake by DCs is affected by several factors. The drainage of antigens into lymph nodes differs between soluble small antigens and particulate large antigens.18) Antigens less than 200 nm in size are readily diffused from injection sites to lymph nodes that typically are rich in DCs, but the uptake of smaller antigens (<5 nm) by DCs is generally inefficient. By contrast, antigens larger than 200 nm in size are not able to enter lymph vessels, but are taken up and transported to lymph nodes by peripheral DCs. Peripheral DCs are localized in peripheral tissue and transfer antigens to cDC1 and cDC2 localized in lymph node.19) Due to the close proximity of cDC2s to lymph sinuses, these cells capture or receive small and particulate antigens and initiate CD4+ T cell responses.20) Otherwise, the penetration of antigens into deep T cell zones is limited, which results in a limited amount of antigen capture by cDC1 and insufficient activation of CD8+ T cells.21) Therefore, an efficient CD8+ T cell response requires a rational strategy for targeting antigens to cDC1.
2.1.2. Specific Receptors for Antigen Uptake in DCsDCs internalize antigens through several mechanisms: clathrin-mediated endocytosis, caveolin-mediated endocytosis, macropinocytosis, and phagocytosis. DCs preferentially take up virus-sized particles (20–200 nm), but macrophage prefer bacteria-sized particles (0.5–5 µm).22) Besides passive uptake, active uptake can be induced using specific ligands for receptors on DCs (Table 1). DCs express a variety of receptors for the recognition and clearance of pathogens. C-type lectin receptors such as mannose receptors, DEC-205, dectin, and DC-SIGN are crucial for the recognition of glycan structures.23) In order to target these receptors, antigens or delivery systems are modified with specific glycans or antibodies. The conjugated vaccines enhance the delivery of antigens to DCs, which leads to robust antigen-specific immune responses compared with non-conjugated vaccines. For example, mannosylated antigens are efficiently internalized in DCs, which allows them to elicit antigen-specific T-cell responses that are 200-fold more efficient compared with that of unconjugated antigens.24) In a similar manner, DCs pulsed with anti-DC-SIGN antibody/antigen conjugates have shown a 100-fold proliferation of peripheral blood lymphocytes.25) DCs also express Fcγ receptors (FcγRs) that bind to the Fc region of immunoglobulin Gs (IgGs). In the presence of specific IgG against antigens, the immune complexes composed of antigens and IgGs are recognized and taken up by the FcγRs on DCs.26) In addition, FcγRs transduce stimulatory signals for the maturation of DCs via an immunoreceptor tyrosine-based activation motif. This type of maturation potentiates DC’s antigen presentation and activation of antigen-specific T cells. Thus, vaccination with antigen-antibody immune complexes not only enhances antigen uptake but it also exerts adjuvant effects. Of note, the expression of these receptors varies in DC subsets, and these receptors are also present on macrophages and other immune cells.27) Therefore, optimal selection of ligands and receptors is required to effectively target DCs.
| Cell type | Receptors | Ligand | Receptor function | |
|---|---|---|---|---|
| Dendritic cells | cDC1 | XCR1 | XCL1 | XCL1 from CD8+ T cells chemoattracts XCR1-espressing cDC1 |
| CLEC9A | F-actin | CLEC9A recognizes F-actin polymers exposed during necrotic cell death | ||
| DEC-205 | Unknown | Endocytic receptor that captures diverse antigens | ||
| BDCA-3 | Thrombin | Anti-coagulation | ||
| cDC2 | CD301b | GalNAc | Endocytic receptor that captures parasite, tumor, and self-glycoproteins | |
| DCIR2 | GlcNAc | Unknown, but immune regulatory role | ||
| BDCA-1 | Glycolipids Acylated peptides | Presents glycolipids and acylated peptides to T cells | ||
| moDC | DC-SIGN | High-mannose or branched fucose glycan | Adhesion, migration, signaling, antigen uptake and presentation | |
| B cells | MZ-B FO-B | BCR | Specific antigen | Signal transduction for antibody production and antigen uptake |
| CR2 | Complement component 3 fragments | Coreceptor of BCR and immune complex uptake High expression in MZ-B | ||
| CD19 | — | Coreceptor of BCR with CR2 and CD81 | ||
B cells exclusively secrete antibodies against pathogens. Mature naïve B cells are classified as either B1 or B2.28) B1 cells are developed in the fetal and early neonatal stages and are maintained by self-renewal, whereas B2 cells originate from bone-marrow stem cells. B2 cells are further divided into follicular B cells (FO-B cells; CD21low and CD23+) and marginal zone B cells (MZ-B cells; CD21high and CD23−). B1 cells and MZ-B cells both rapidly secrete antibodies in a T-cell independent manner in response to bacterial saccharides, self-antigens, and artificial polymers.29,30) On the other hand, FO-B cells secrete antibodies against countless proteinous antigens with the help of T cells in delayed stages, but they secrete high-affinity and class-switched antibodies following the formation of germinal centers.31) B1 cells and MZ-B cells are predominantly localized in the peritoneal cavity and spleen, respectively, but FO-B cells are distributed in systemic lymph tissues.28)
In lymph nodes and in the spleen, B cells organize a B-cell area (follicle) that is separated from the T-cell zone (Fig. 1). MZ-B cells rapidly and directly encounter blood-borne antigens because blood flow first reaches the marginal zone (MZ) in the spleen, while FO-B cells recognize antigens in B-cell follicles.32) Smaller proteins (<70 kDa) directly access follicles through a conduit system, but larger particles must be transferred to follicles either by MZ-resident cells in the spleen or by subcapsular macrophages in lymph nodes.33,34) The transferred antigen particles are displayed on follicular DCs, and are then recognized by FO-B cells.
B cells share some features with DCs in terms of APCs, which includes high expressions of MHCs, costimulatory molecules, and production of cytokines.35) Antigen presentation on MHC class 2 by antigen-specific B cells is well known to stimulate the proliferation of cognate CD4+ T cells. After priming antigen-specific B cells with antigens, the B cells migrate to the T–B border, activate cognate CD4+ T cells, and trigger germinal center formation to produce high-affinity antibodies. Although the contribution of B cells to MHC class 1-mediated CD8+ T cell induction remains controversial,36) several groups have reported that B cells exhibit cross-presentation ability and elicit CD8+ T cell responses.15,37,38) The cross-presentation pathway in B cells is not completely elucidated, but B cells are known to cross-present antigens by utilizing autophagy and proteasome-dependent pathways following the internalization of exogenous antigens.39) Similar to DCs, antigen presentation ability differs according to the B-cell subset. Compared with FO-B cells, MZ-B cells are known to express high amounts of MHC and co-stimulatory molecules,40) which makes MZ-B cells attractive APC candidates.
2.2.2. Specific Receptors for Antigen Uptake in B CellsPrimary antigen uptake by B cells is mediated by BCRs in an antigen-specific manner. BCRs with a high affinity against specific antigens exhibit a drastic reduction in the antigen dosage required for antigen presentation and T cell activation.41) Following the internalization of specific antigens, B cells present antigen-derived peptides and efficiently expand the clonal proliferation of cognate T cells. In fact, the adoptive transfer of antigen-specific B cells pulsed with antigens tends to proliferate cognate T cells more efficiently than that of non-specific B cells.42,43) However, the number of antigen-specific B cells is usually very low (<0.1%)44,45) so the total antigen presentation activity would also be low if the number of antigen-specific B cells is not increased. Other approaches to enhance antigen uptake are the utilization of some receptors like FcγRs and complement receptors that are also expressed in DCs (Table 1). One of the B cell-specific receptors, complement receptor 2 (CR2 or CD21), recognizes fragments of C3.46) Macrophages and DCs express similar receptors, namely complement receptors 3 (CR3 or CD11b) and 4 (CR4 or CD11c). The moieties of C3 fragments recognized by CR differ: iC3b for CR2, CR3 and CR4; and, C3d for CR2.46) Therefore, conjugation of C3d or anti-CR2 antibody achieved a specific targeting of antigens to B cells and showed an enhancement of vaccination efficacy.47,48) In addition, FcγR2b is mainly expressed on B cells, although other FcγRs (FcγR1, FcγR2a, FcγR3, and FcγR4) are expressed on macrophages and DCs. However, FcγR2b has an immunoreceptor tyrosine-based inhibitory motif,49) so the targeting of FcγR2b might attenuate immune responses against antigens.
Passive targeting of antigens to DCs is primarily performed using nanoparticles. Nanoparticles such as liposomes, virus-like particles, and polymer nanoparticles are passively internalized by DCs owing to their phagocytic activity. Therefore, nanoparticle platforms have been applied in the development of vaccines.12) Liposomes comprise the most promising nanoparticle systems used for antigen delivery due to their biocompatibility and versatility in the loading of antigens.50) Liposomes can stably encapsulate and deliver antigens and adjuvants to lymph tissues, which enhances both humoral and cellular immune responses. In addition, manipulation of their sizes and charges improves their drainage to lymph nodes and their uptake by APCs.51) Recently, WHO recommended the application of liposome-based malaria vaccines, RTS, S/AS01, for children in Sub-Saharan Africa and in other regions. To produce this vaccine, malaria sporozoite antigens were formulated together with a liposome-based adjuvant (AS01; monophosphoryl lipid A and QS-21). Immunization with AS01 was confirmed to elicit multifunctional antibodies to malaria.52) The effect of AS01 was not presumably due to an increase in the antigen uptake by DCs, but, rather, to the upregulation of co-stimulatory molecules and to the recruitment of DCs in draining lymph nodes.53) Another application example is that of liposome-based vaccines in the treatment of cancers. Stimuvax is one of the liposomal cancer vaccines that contains Mucin1 (MUC1) glycoproteins formulated with liposomes and adjuvants (monophosphoryl lipid A and BLP25).54) Although pre-clinical and phase 1 studies have demonstrated that Stimuvax exhibits safety and immunogenicity, phase 2/3 trials failed to prolong overall survival for patients with non-small cell lung cancer.55) Subgroup analyses indicated that the combination of radiology and chemotherapy prolonged the survival of patients, so further studies were conducted with other therapeutics.
In order to enhance the therapeutic efficacy of these vaccines, many researchers have also utilized active-targeting strategies. Conjugation of specific ligands and antibodies allows active targeting to receptors on DCs.23,56) Interactions of ligands or antibodies with receptors leads to the internalization of antigens and presentation onto MHC molecules in addition to an upregulation of stimulatory signals. To date, hundreds of DC-targeted vaccines have been studied in the pre-clinical stage and some of them have been entered into clinical trials (Table 2). One of the most famous approaches for targeting to DCs is the use of specific antibody against DEC-205, which is confirmed to express in mouse and human DCs. CDX-1401 (Table 2) is one of the cancer vaccines composed of NY-ESO-1 (cancer testis antigen) fused with human anti-DEC-205 antibody and adjuvant that has been injected in patients with cancers in phase 1 clinical trials.57) Both antibody and T cell responses against NY-ESO-1 were induced and half of the patients tested showed stable diseases. CDX-1307 (Table 2) is another type of DC-targeted vaccine composed of hCG-β (human chorionic gonadotropin) fused with anti-mannose receptor antibodies. The phase 1 clinical trial of CDX-1307 showed the induction of anti-hCG-β antibodies and hCG-β-specific T cells.58) However, active targeting to DCs does not always lead to clinical trial success. Lipovaxin is a DC-SIGN-targeted vaccine that is prepared via the fusion of melanoma-derived cell membrane vesicles with phosphatidylcholine-based liposomes and decoration with an immunoglobulin single-variable domain against DC-SIGN.59) Lipovaxin exhibited antitumor effects against melanoma in a pre-clinical mouse model, but unfortunately failed to induce antitumor immune responses in phase 1 clinical trials.60)
| Category | Disease | Therapy | Phase | Ref |
|---|---|---|---|---|
| In vivo dendritic cell-targeted vaccine | Stage IIB-IV melanoma | Anti-DEC-205 antibody/NY-ESO-1 fusion protein with Poly: ICLC and Flt3-ligand, CDX-1401 | 2 | NCT 02129075 |
| Bladder cancer | Mannose receptor-targeted hCG-β vaccine, CDX-1307 | 2 | NCT 01094496 | |
| Melanoma | DC-SIGN–targeted lentiviral vector encoding NY-ESO-1 with pembrolizumab | 1 | NCT 02122861 | |
| HIV | Anti-DEC-205 antibody/HIV gag p24 fusion protein with Poly: ICLC | 1 | NCT 01127464 | |
| Ex vivo dendritic cell-based vaccine | Non-Hodgkin lymphoma | Dendritic cells exposed to dead fragments of lymphoma cells | 3 | NCT 00006434 |
| Recurrent prostate cancer | Alpha-type 1 dendritic cells loaded with allogeneic prostate cell lines with androgen ablation | 2 | NCT 00970203 | |
| Recurrent grade III and IV Brain Tumors | Dendritic cells pulsed with CMV pp65-LAMP mRNA with nivolumab | 1 | NCT 02529072 | |
| Chronic HCV-infection | Dendritic cells pulsed with recombinant HCV Core and NS3 proteins | 1/2 | NCT 03119025 | |
| Ex vivo B cell-based vaccines | B-chronic lymphocytic leukemia | B-chronic lymphocytic leukemia transduced with CD40L and IL-2 | 1 | NCT 00458679 |
| Uterine cervical neoplasm | Autologous B cells and monocytes transfected with E6E7 gene of HPV | 1/2 | NCT 02866006 |
Some aspects of the successes of DC-targeted vaccines warrant further attention. One is the expression pattern of specific receptors in different DC subsets, which determines the polarization of immune responses. CLEC9a is dominantly expressed in cDC1, and thus targeting antigens to CLEC9a induces a robust CD8+ T-cell response in mice.61) On the other hand, dectin-1 is expressed in mouse cDC2, so targeting dectin-1 induces CD4+ T-cell and antibody responses rather than CD8+ T cell responses.62) In addition, the expression pattern differs between humans and mice.56) The expression of DEC-205 has been confirmed in different types of human DCs, such as cDC1, cDC2, and pDC. In contrast, the expression of mannose receptors and DC-SIGN in humans is limited to monocyte-derived DCs that are atypical. Therefore, targeting DEC-205 is likely to be the most promising approach, although DEC-205-targeted vaccines require adjuvants in order to avoid immune tolerance. From another point of view, the selection of either ligand or antibody should be considered.63) Antibodies possess high affinity with specific receptors, which enables them to target a specific subset of DCs, albeit with a high cost for manufacturing and concerns of anti-drug antibody induction.64) Some ligands such as glycans are easily manufactured and conjugated to increase affinity.65) Glycans are recognized by multiple receptors in DCs, which could enhance the efficiency of their delivery to DCs and the subsequent immune responses. Accumulating evidence of such aspects is expected to lead to the clinical success of DC-targeted vaccines in the future.
3.2. B Cell-Targeted VaccinesAlthough there are few reports on B cell-targeted vaccines, these cells have a unique potential to elicit immune responses. In particular, they represent a rational approach to the induction of humoral immune responses owing to their exclusive properties of antibody production.66) Antigen-specific B cells produce antibodies after the recognition of antigens by BCRs with the help of cognate T cells. Accordingly, antigens themselves can be targeted to antigen-specific B cells. Moreover, surface decoration of nanoparticles with multivalent antigens increases both their affinity and the binding to BCRs, which has resulted in robust antibody production in mice.67) Indeed, amyloid beta peptide-decorated liposomes with adjuvants, which are vaccines for the treatment of Alzheimer diseases, have induced the production of anti-amyloid beta antibodies in mice.68) In addition to the increased uptake of antigen-decorated nanoparticles, they also have the ability to cross-link BCRs on B cell surfaces, which enables the B cells to induce antibodies without the help of T cells. However, as mentioned above, the population of antigen-specific B cells in the body is normally very low, which unfortunately means the antigen presentation to non-cognate T cells is insufficient.
B cell-specific molecules have been explored for their ability to target whole B cells. The targeting of B cell-specific molecules would be achieved without concern for antigen specificity. CD19 is a universal B cell marker in all B cell lineages and is an aid to BCR signaling. In previous study, antigen/anti-CD19 antibody conjugates were taken up by splenic B cells and in turn antigen-specific CD4+ T cells were activated in mice.69) Additional studies have shown that the conjugation of MUC1 (tumor-associated antigen) with anti-CD19 antibody elicits not only CD4+ T cell responses, but also a high titer of anti-MUC1 antibodies and CD8+ T cell responses.70) Finally, the conjugates mixed with CpG adjuvant have suppressed the growth of tumors, suggesting that antigen delivery to B cells is a promising approach to induce antitumor immune responses. CD23 (low affinity receptor for IgE) and FcγR2b (receptor for IgG) are also known to express in mature B cells. So, conjugates composed of antigen/IgE and antigen/IgG are taken up by B cells through CD23 and FcγR, respectively. In fact, the administration of ovalbumin (OVA)/anti-OVA IgE conjugates has enhanced OVA-specific antibody production and CD4+ T cell activation compared with the administration of OVA alone in mice.71) On the other hand, OVA/anti-OVA IgG2a conjugates have definitely enhanced OVA-specific antibody production and CD4+ T cell activation in wild type mice. Interestingly, in FcγR2b-knockout mice, the immune responses were further enhanced, suggesting that the vaccination effect of OVA/anti-OVA IgG2a conjugates were downregulated by FcγR2b that possesses an immunoreceptor tyrosine-based inhibitory motif.49) It is estimated that OVA/anti-OVA IgG2a conjugates are taken up by not only FcγR2b on B cells but also by other FcγRs on B cells and on other cells, and that enhanced immune responses could be attributed to other types of FcγR expressed in B cells and other cells, but not the FcγR2b expressed in B cells.
It is unclear whether the utility of B cell-targeted vaccines is superior to that of DC-targeted vaccines. Treatment involving a combination of both vaccines could increase their effect. Immune-stimulating complexes (ISCOM), involve well-studied nanoparticles consisting of saponin adjuvant (Quil A), phospholipids, and cholesterol that can be taken up by DCs.72) Incorporation of CTA1-DD adjuvant, in which DD is from the protein A domain, into the ISCOM formulation enables the targeting of B cells through DD, which binds to Ig receptors in the B cells. The combined targeting ability to both DCs and B cells synergistically results in enhanced induction of antibodies and CD4+ T-cell responses.73) In addition, study into DC-derived exosome vaccines has revealed the crucial role that B cells play in the induction of antibodies and in CD4+ T-cell and CD8+ T-cell responses.74) Immunization with antigen-loaded DC exosomes elicits the proliferation of CD4+ and CD8+ T cells in wild type mice, but the proliferation proved to be defective in B cell-deficient mice. The collaboration of B cells and DCs appears to maximize vaccination effects. Another unexplained question is which B cell subsets contribute to the induction of immune responses. As mentioned above, MZ-B cells produce antibodies more rapidly, present antigens more efficiently, and activate T cells due to high expression of MHC and costimulatory molecules.40) Accordingly, MZ-B cells are assumed to be the most suitable target B cells to deliver antigen in order to induce strong immune responses. However, MZ-B cell-targeted vaccines have yet to be developed.
As another potential method for immunization, cell-based vaccines have been investigated in animal models and clinical trials.75) In cell-based vaccines, APCs or their precursors are isolated from a patient’s body and differentiated into mature APCs using appropriate stimulating factors such as recombinant cytokines and TLR agonists. The mature APCs then are loaded with antigens and infused back into the patient’s body whereupon they induce immune responses. Antigens can definitely be loaded to APCs without being affected by an immune-suppressive environment such as that created by diseases such as cancer.76) This makes cell-based vaccines an effective approach, and promising results have been shown in several clinical trials57) (Table 2). The efficacy of DC-based vaccines have been reviewed elsewhere, and the following section is focused mainly on the construction of DC-based vaccines to improve their efficacy.
DCs are promising APCs that are the most commonly used vehicle for cell-based vaccines in preclinical animal models as well as in clinics. Actually, one DC-based vaccine, Sipuleucel-T (PROVENGE®, Dendron), which is composed of monocyte-derived DCs loaded with the tumor antigen PA2024 (a complex of prostatic acid phosphatase fused with granulocyte-macrophage colony-stimulating factor (GM-CSF)), has been approved for the treatment of castration-resistant prostate cancer,77) although its clinical responses have been limited when it is used as a monotherapy. A number of studies have been conducted to develop more prominent DC-based vaccines by maximizing the advantages of DCs.
One distinct feature of DCs is that they are composed of several subsets, and each of these possess different progenitors, functions and migration patterns.17) DCs that differentiate from either CD14+ peripheral blood monocytes78) or the CD34+ hematopoietic precursor cells isolated from a patient’s blood79) have been widely used for research, and analysis has revealed that ex vivo-induced DCs are more similar to macrophages than to DCs induced in vivo.80) Therefore, researchers have attempted to develop methods for differentiating DCs in order to reveal those that express characteristics similar to the in vivo version. Kirkling et al. developed a method for generating DCs that closely resemble in vivo cDC1 by culturing DC progenitor cells with OP9 stromal cells expressing the Notch ligand Delta-like 1, which has the ability to send important signaling that allows cDC1 differentiation. Such differentiated cDC1s have successfully induced antigen-specific CD8+ T cell responses after being loaded with antigens and infused into animals.81) Differentiation of XCR1+ DCs that possess the useful ability to cross-present could be achieved by culturing human CD34+ hematopoietic cells with recombinant human cytokines FLT3-L, SCF, GM-CSF, and interleukin-4 (IL-4).82) These in vivo like DCs would aid in the development of effective DC-based immunotherapies by demonstrating the effectiveness of their antigen-presenting capacity.
Another distinct feature of DCs is that they have the ability to take up extracellular antigens and then present them not only to CD4+ T cells but also to CD8+ T cells via cross-presentation. DCs can take up various antigens including antigen-encapsulating nanocarriers such as liposomes84) and PLGA nanoparticles.85) Those nanocarriers perform not only as an antigen delivery system to DCs but also as a stimulator of DCs to enhance the expressions of MHCs and co-stimulatory molecules such as CD80 and CD86.85) We may be able to simplify the process of DC stimulation and antigen loading to DCs by using a nanocarrier system.
Although DCs are capable APCs, one drawback exists; DCs poorly migrate to lymphoid tissues such as lymph nodes and spleen after injection by any route. Therefore, the development of cell-based vaccines that use DCs requires improvement in migration ability. Accordingly, several approaches have been used to overcome the poor migration ability of DC’s, which has enhanced the immune response. Because DCs express CCR7, they have a unique property that allows them to home in to lymphoid tissues in response to CCL21 and CCL19 gradients, which are produced in high endothelial venules and T cell zone.86) Accordingly, the additional transduction of CCR7 genes into DCs greatly enhances their homing ability to lymph nodes, which results in increased T cell responses following intradermal injections of DCs.87) In addition, when DCs are transduced with factors such as cytoskeleton proteins, their abilities for lymph node migration and immune-response induction are improved.88) There is another strategy that utilizes magnets to attract DCs to lymph nodes.89) In this strategy, DCs loaded with nanoparticles that contain antigen peptides and iron oxides are injected into mice wearing a magnetic ring, which provides a magnetic pulling force. The magnetic pull enriches the lymph nodes near the magnetic ring with nanoparticle-loaded DCs, and this has augmented the anti-tumor immunity of DCs in animal models.
4.2. Ex Vivo B Cell-Based VaccinesAlthough DCs are now widely used in the field of cell-based vaccines, as introduced above, B cells have also been used as alternative APCs (Table 2) because B cells were considered major APCs before the discovery of DCs. However, B cells have a relatively lower capacity for antigen uptake and presentation compared with that of DCs.90) Thus, vaccine designs using B cells must either overcome the shortcomings of B cells or find a way to maximize the advantageous features of B cells.
One of the most significant advantages of B cells is that they amount to the largest possible population for APCs.91) Therefore, large numbers of B cells can be easily separated and obtained from blood. In addition, there is no need for further differentiation, which is not the case when using DCs. In some studies, antigen-loaded B cells were prepared and administered to cancer-model animals within 24 h after isolation from the body and consequently began to induce antitumor immune responses.92) There is another approach to increasing the number of B cells in vitro after isolation from the body. This refers to a culture system that utilizes CD40L, which is the protein expressed by the activated CD4+ T cells in the body that stimulates naïve B cells. The proliferation of B cells can be activated using recombinant soluble CD40L or feeder cells expressing CD40L in vitro. The resultant “CD40-activated B cells” express high levels of the MHC molecules CD80 and CD86,90) and these B cells then efficiently induce antigen-specific immune responses.
The ability of B cells to phagocytose extracellular antigens is low by comparison with that of DCs. Although B cells are able to take up antigen through antigen-specific BCRs, the population of antigen-specific B cells is usually very small. Therefore, the antigen-loading method, which loads antigens onto B cells, must be well designed. The most common approach is the use of MHC-restricted antigen peptides.92) These peptides can be loaded to the MHC molecules expressed on B cell surfaces without internalization. The loaded antigens are presented to T cells followed by the activation of T cells. However, this method is restricted to peptide antigens, and cannot be applied to proteins or mRNAs. Another method involves targeting the complement receptors (CRs) and CD19 of B cells. Complement-activating liposomes contain antigens, which were internalized via CR-mediated endocytosis,93) or anti-CD19 antibodies conjugated with antigens,94) which were internalized via CD19-mediated endocytosis. Furthermore, B cells that express antigen-specific BCRs have also recently been used for B cell-based vaccines.95) Wennhold et al. purified OVA-specific B cells using antigen-biotin tetramers from donor mice pre-immunized by OVA and activated them using CD40L. Those B cells are able to internalize antigen via BCRs to not only induce OVA-specific T cell responses, but also to secrete antibodies against OVA following infusion to recipient mice.
B cells have the ability to migrate into either the spleen or lymph nodes. In particular, B cells with a high expression of CXCR5 migrate in response to the gradient of their ligand CXCL13, which is produced in follicles.96) In addition, naïve B cells originally patrol lymphoid tissues and blood, so transplanted (ex vivo) antigen-loaded B cells can also migrate to lymphoid tissues after their injection. One study compared the migration of injected antigen-loaded B cells and antigen-loaded DCs, and a larger number of B cells were observed in both spleen and lymph nodes.97) B cells home-in on follicular regions located next to the T cell zones in the spleen. Zhang et al. devised a strategy for B cell-based vaccines on the basis of the migration pattern of transplanted B cells.97) They focused on central memory T cells (TCM), which exist in the T cell zones and immediately respond to a second infection. Thus, they injected viral vector-loaded B cells for booster vaccines. After injection, B cells expressing antigen protein accumulated in the follicular region in the spleen and lymph nodes and efficiently boosted the antigen-specific T cell responses that resulted from the activation of TCM by the B cells.
Thus far, we have summarized the current reports on DC- or B cell-targeted/based vaccines. Regarding DC vaccines, the targeting of antigens to specific DC subsets such as cDC1 and cDC2 has been well investigated, but antigen delivery to specific B-cell subsets such as MZ-B and FO-B has never been reported. In order to improve the efficacy of B cell-targeted vaccines, further selective antigen delivery to specific B cell subsets is required. As mentioned above, MZ-B cells could be one of the promising candidates among APCs due to significant levels of antigen presentation and expression of co-stimulatory molecules. However, there has been no report on antigen delivery to splenic MZ-B cells. We previously developed a method to deliver antigens to MZ-B cells by repeat intravenous (i.v.) injections of PEGylated liposomes. We found that the second dose of PEGylated liposomes is selectively taken up by splenic MZ-B cells.98) In this section, we provide details of the splenic MZ-B cell-targeted vaccines we developed (Fig. 2).
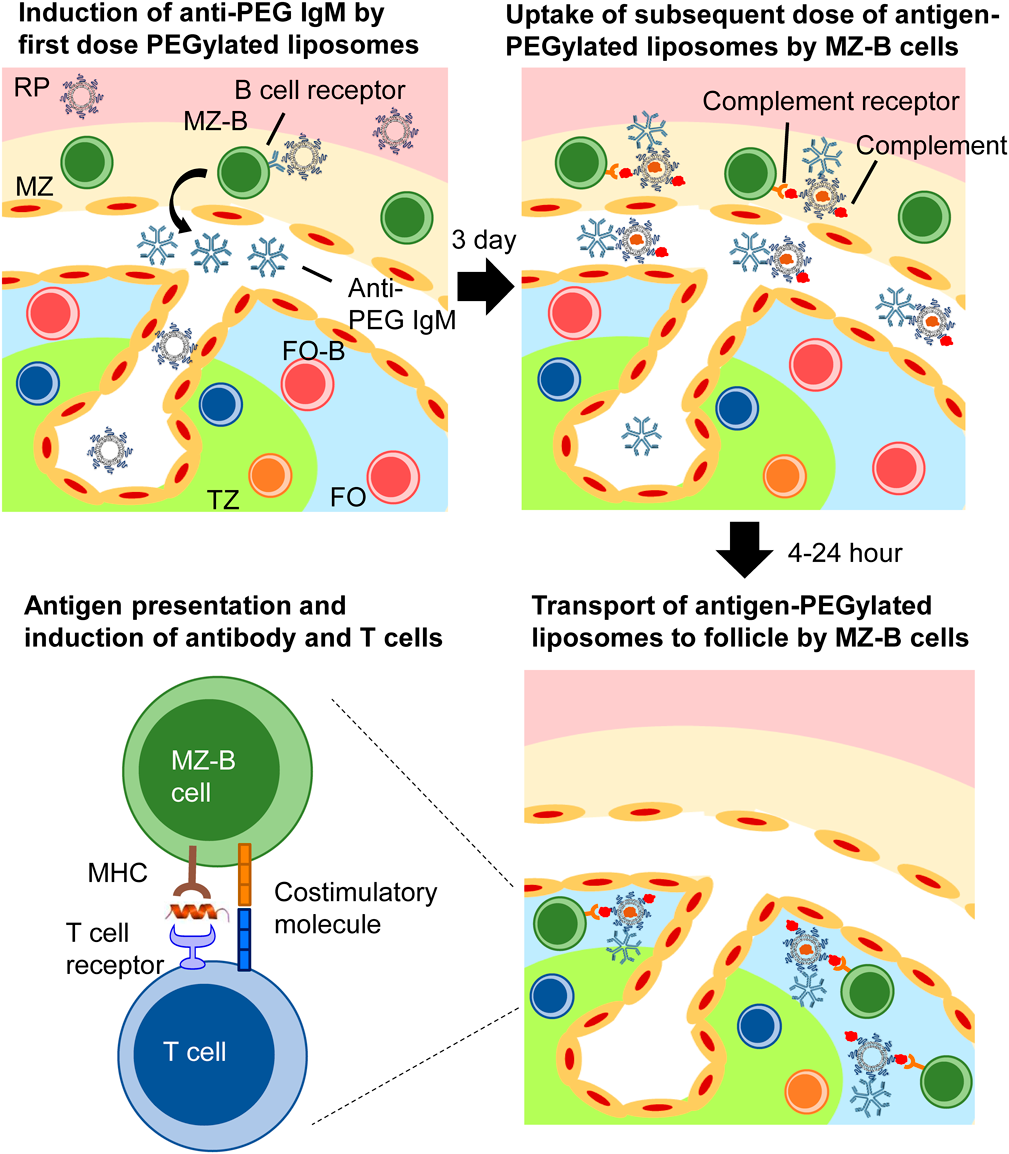
PEGylation is a well-known technology for extension of the in vivo half-life of protein drugs or nanocarriers after injection. At this point, more than 20 PEGylated therapeutics are clinically available.99) PEGylated liposome is on the list of clinically successful nanocarriers. PEGylated liposomes are also used in the field of vaccines for the purpose of antigen delivery in preclinical animal studies.100,101) In the case of subcutaneous (s.c.) injection of liposomes, PEGylation is known to enhance the drainage of liposomes to lymph nodes by inhibiting interactions with cells at the injection sites.102) Nevertheless, such a stealth effect of PEGylation impaired the uptake of liposomes by the APCs in lymph nodes. In a similar manner, i.v.-injected liposomes are usually taken up by macrophages in the liver and spleen, but PEGylation decreases such aggressive uptake, which results in decreases in the liposome uptake by APCs in the liver and spleen.
In our previous studies, we demonstrated that i.v. injections of tiny dose of PEGylated liposomes elicits the production of anti-PEG IgM,103–105) and a subsequent dose injected on Day 3 accumulated in the spleen rather than in the liver.98) Further study indicated that PEGylated liposomes that accumulated in the spleen were taken up mainly by MZ-B cells with the help of bound anti-PEG IgM and subsequent complement activation. Binding of anti-PEG IgM to PEGylated liposomes is known to activate complement systems104) Thus, the PEGylated liposomes are decorated with C3 fragments.106) Generally, C3 fragments are recognized by CR2 on MZ-B cells, which are expressed at a higher level in MZ-B cells than in FO-B cells.32) Consequently, the second dose of PEGylated liposomes was selectively taken up by the MZ-B cells rather than by FO-B cells under our experimental conditions.
5.2. Enhancement of Humoral and Cellular Immune Responses by Delivery of Antigens to Splenic MZ-B Cells with PEGylated LiposomesInterestingly, further observation showed that following the capture of PEGylated liposomes, MZ-B cells transport the captured PEGylated liposomes to follicles in a time-dependent manner.98) Follicles are adjacent to the T-cell zones and to the sites of immune response maturation such as affinity maturation and the class switching of antibodies.31) Therefore, we hypothesized that follicular antigen delivery with PEGylated liposomes must enhance antibody production. OVA was encapsulated in the second dose of PEGylated liposomes for antigen delivery. Following a first i.v. injection of empty PEGylated liposomes for priming the immune system, a second dose was given. Consequently, the second dose enhanced the production of anti-OVA antibodies in mice and rats.107) The enhanced specific antibody production was cancelled by treatment with FTY720, which suppresses MZ-B cell-mediated follicular transport.108) In addition, s.c. immunization with empty PEGylated liposomes and OVA-loaded PEGylated liposomes induced few antibodies probably due to a lack of drainage to the spleen. These findings suggest that antigen delivery to splenic MZ-B cells and follicles, via repeated i.v. injection of PEGylated liposomes, is critical for the enhancement of specific antibody production and becomes an alternative immunization method, which will be useful in the design of new vaccines.
Our unique antigen delivery system for splenic MZ B cells uses repeated i.v. injections with PEGylated liposomes to elicit antigen-specific CD8+ T cells that can mediate antitumor immune responses.37) Although a single immunization combined with empty PEGylated liposomes and OVA-loaded PEGylated liposomes could not induce cytotoxic T lymphocytes (CTLs), a three-fold regimen of immunization successfully induced CTLs. In addition, incorporation of α-galactosylceramide (adjuvants), which is an activator of natural killer T cells,109) into OVA-loaded PEGylated liposomes decreased the required number of immunizations for activation. Single immunization with only PEGylated liposomes co-encapsulated with antigens and adjuvants induced CTLs and natural killer T cells. A single immunization with a type of anti-tumor vaccine induced CTL, which resulted not only in prevention, but also in suppression of the growth of OVA-expressing tumors in vivo.37) These results indicate that splenic MZ-B cells play a key role in the induction of antigen-specific CD8+ T cells as APCs in our immunization system. Further study confirmed that MZ-B cells, but not FO-B cells, presented antigen-derived peptides onto MHC class 1 molecules.37) In addition, the adoptive transfer of antigen-presenting MZ-B cells that is separated from the spleen and loaded antigens in vitro increased the number of antigen-specific CTL in recipient mice. Accordingly, antigen delivery to splenic MZ-B cells could become an effective approach to vaccination for the induction of both humoral and cellular immune responses.
5.3. Next-Generation Approach for Antigen Delivery to Splenic MZ-B CellsOur immunization approach requires a priming process using i.v. injection of empty PEGylated liposomes before i.v. injection with antigen-loaded PEGylated liposomes, which is complicated and time-consuming. In this system, it is assumed that the second dose of antigen-loaded PEGylated liposomes will be recognized by anti-PEG IgM induced by the first dose, and then opsonized by C3 in blood circulation and finally taken up by MZ-B cells through CR2.110) We therefore assumed that the PEGylated liposomes, if they could be directly opsonized by C3, would be taken up by MZ-B cells without priming with a first dose of empty PEGylated liposomes. C3 is known to react with the amine and hydroxyl groups of pathogens in their activation process.106) Although traditional PEGylated liposomes with methoxy groups at the end of PEG scarcely activate complement systems, hydroxy PEG-modified liposomes are spontaneously opsonized by C3. Therefore, such PEGylated liposomes decorated with C3 fragments might be taken up by splenic MZ-B cells even after a single injection.93) Optimization studies of PEG density have shown that 2% hydroxy PEG-modified liposomes are the most effective to increase the uptake by MZ-B cells compared with 0.5 or 5% hydroxy PEG-modified liposomes. Finally, OVA-loaded hydroxy PEG (2%)-modified liposomes induced the production of anti-OVA IgG and also induced the activation of OVA specific T cell without priming using empty PEGylated liposomes.93) Therefore, this novel delivery approach via the use of hydroxy PEG (2%)-modified liposomes is a simple and efficient method for the induction of meaningful levels of humoral and cellular immune responses.
Targeting antigens to APCs is essential to enhance vaccination efficacy while decreasing adverse effects. To date, countless studies have been conducted in an effort to develop vaccines targeting or utilizing DCs that are professional APCs and initiators of various types of immune responses. However, unique characteristics, such as antigen-specific uptake, high migration ability, and ease of ex vivo culture and proliferation, make B cells attractive targets for vaccines. Effective induction of immune responses against antigens could be possible via the targeting or utilization of B cells. Similar to DCs, optimal B cell subset selection is one of the obstacles in the development of efficient B cell-targeted vaccines. Our recent study demonstrated, however, that specific B cell subsets such as splenic MZ-B cells are promising for enhancing vaccination effects. In the future, it is expected that B cell-targeted/based vaccines could become an alternative or a compensating regimen in combination with DC-targeted/based vaccines.
This study was in part supported by a Grant-in-Aid for Young Scientists (15K18921), a Grant-in-Aid for Transformative Research Areas (A) (Publicly Offered Research) (21H05526), by the Takeda Science Foundation, by the Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan and by a research program for the development of an intelligent Tokushima artificial exosome (iTEX) from Tokushima University. The authors are grateful to Mr. James L. McDonald for his helpful advice in developing the English manuscript.
The authors declare no conflict of interest.