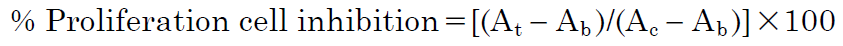Abstract
Two series of 2-substituted benzimidazole conjugated 1,3,4-oxadiazole derivatives were designed, synthesized and evaluated for their cytotoxic activities against the three human cancer cell lines (cervical cancer (HeLa), breast cancer (MCF-7) and lung cancer (A549)). As the results 14 compounds demonstrated consistent to stronger cytotoxicities compared to the control 5-fluorouracil (5-FU) towards the tested cell lines including 4c (HeLa); 4b, 4e, 4h, 7i–j, 7m–n, 7s (MCF-7); 7b (MCF-7, A549); 7h (HeLa, MCF-7); and 4d, 4i, 7c (HeLa, MCF-7, A549), with the IC50 ranging from 2.7 to 38 µM. Notably, compound 4b illustrated almost 5-fold activity against the MCF-7 while 4d, 4i were 9- and 8-fold (HeLa), 4.5- and 13-fold (MCF-7), 4.7- and 4-fold (A549) increase in activity compared to 5-FU, respectively, and were found as lead compounds. These findings suggest that compounds 4b, 4d and 4i merit further characterization and can serve as promising scaffolds in the discovery of new potent anticancer agents.
Introduction
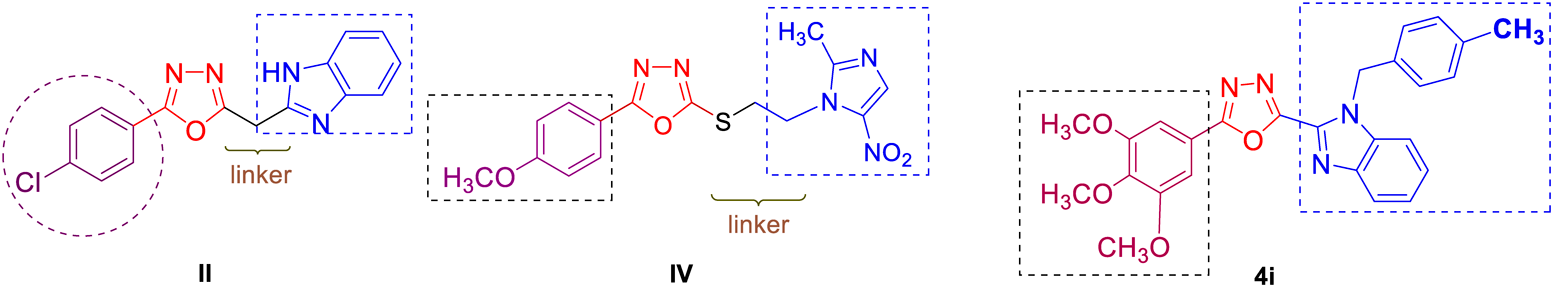
Nitrogen and oxygen containing heterocyclic compounds have been used as a scaffold to synthesize numerous therapeutic molecules.1–5) Among such privileged moieties, 1,3,4-oxadiazoles6–10) and benzimidazole11) have attracted wide attention due to their useful biological properties. 1,3,4-Oxadiazoles are very good bioisostere of amides and esters, which can contribute substantially in increasing pharmacological activity by participating in hydrogen bonding interactions with the receptors.12) In recent years, extensive research has focused on developing novel 1,3,4-oxadiazole-heterocycle hybrids as anti-tumor drugs since these structures have been proven to overcome various disadvantages of current anticancer drugs such as drug resistance, toxicity, and other side effects.13) On the other hand, benzimidazole, the isostere of a purine-based nucleic acid with desirable electron-rich characteristic which is beneficial for binding with a variety of therapeutic targets, is one of the most prominent heterocycle with cytotoxic properties against different types of cancer cell lines.14–17) Literature survey revealed that benzimidazole conjugated 1,3,4-oxadiazoles showed remarkable anticancer activity. Compounds I18) and II19) in Fig. 1 are such representative examples, which share a common structural characteristic of containing the free N–H position on the benzimidazole heterocycle while 1,2-disubstituted benzimidazoles have recently been reported to possess novel cytotoxicity against cancer cell lines.14,15,20) Furthermore, while there have been several reported anticancer oxadiazole/benzimidazole hybrids which rely on the use of a linker such as an alkyl chain as in the case of I and II or an arene ring,21,22) no example on the anticancer activity of 1,3,4-oxadiazole linked directly to the C-2 position of the benzimidazole ring. We reasoned that direct combination of 1,3,4-oxadiazole and N-substituted benzimidazole scaffolds in a single molecule of series A (Fig. 1) would lead to novel structures with potential cytotoxic agents.
On the other hand, upon literature survey we found that oxadiazole derivatives IIIa–b bearing the methylthioether linkage showed stronger cytotoxicity against MCF-7.23) Similarly, compound IV24) exhibited as a potential anticancer agents against HepG2, SGC-7901, and MCF-7 cell lines. Noticeably, Mochona reported the synthesis and anticancer activity of hybrid oxadiazole derivatives V with the benzimidazole scaffold incorporated through the thioether linkage.25) However, only compound Vc (R = F) exhibited moderate cytotoxicity against MDAMB-231 breast cancer cell lines (IC50 > 17.5 µM). Based on the fact that most biologically active benzimidazole based compounds bearing functional groups at the 1, 2, and/or 5/6 positions,26) further introduction of different substituents on the two benzene rings of the benzimidazole moiety of compound V would consequently increase the activity.
Based on the above findings and also in the continuation of our efforts to explore the anticancer potential of heterocycles,27–29) herein we report the design and synthesis of two series of hybrids by combining the 1,2-disubstituted benzimidazole pharmacophore and (i) 1,3,4-oxadiazole (series A, Fig. 1) and (ii) thioether linked 1,3,4-oxadiazole (series B) in single molecular framework and their in vitro cytotoxicity evaluation against the three human cancer cell lines (HeLa, MCF-7 and A549).
Results and Discussion
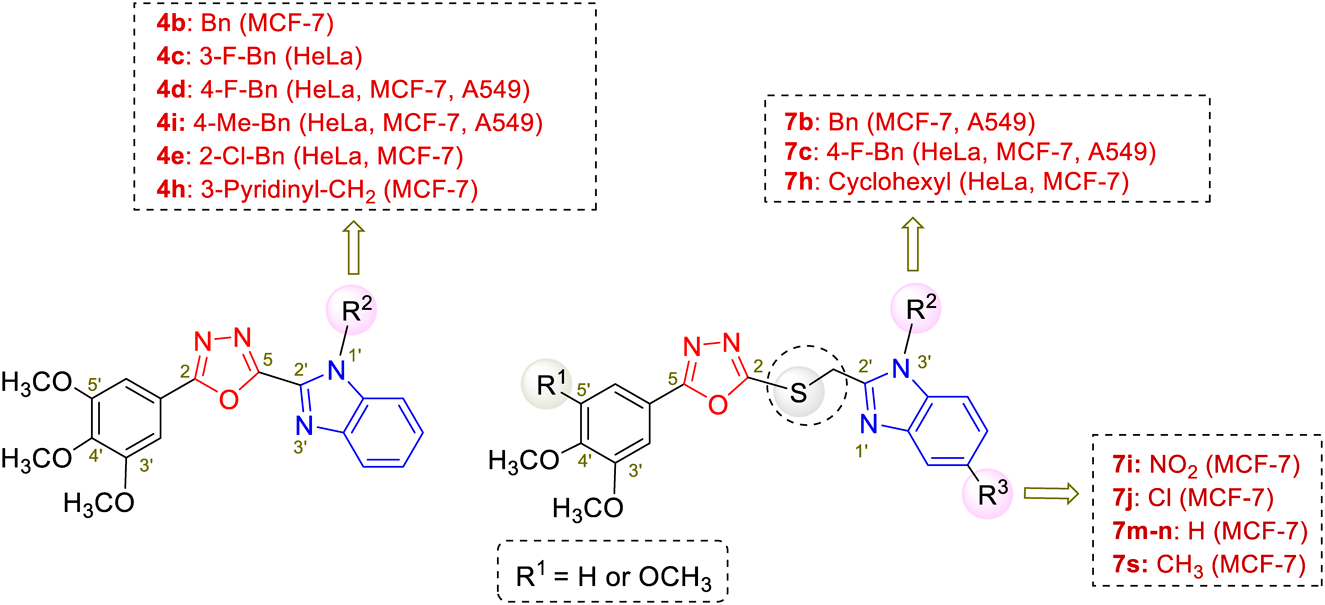
ChemistryTo search the structure and activity relationship analysis (SAR) of hybrid structures, we designed two series of hybrids as depicted in Chart 1.

The design made use of directly incorporating the benzimidazole moiety to the C-5 position of the 1,3,4-oxadiazole (compounds 4a–q) or through a methylthioether linkage (compounds 7a–s). The methoxy substituted phenyl moieties were chosen to install at the C-2 position of the 1,3,4-oxadiazole rings of the two series based on literature reports30–33) as well as our previously observed results27,28) with respect to this structural feature in enhancing the cytotoxicity. Starting from the commercially available substituted ethyl benzoates 1, aminolysis of the ester moieties took place using hydrated hydrazine in ethanol as the solvent to provide the corresponding benzohydrazide derivatives 2, which were subsequently condensed with different aromatic aldehydes 334,35) using iodine as the oxidant under mild basic conditions (K2CO3)36) to complete the desired 1,3,4-oxadiazole derivatives (4a–q) in good total yields (69–85%) (Table 1). For the synthesis of the hybrid 1,3,4-oxadiazole-benzimidazole derivatives (7a–s) (Chart 1), benzohydrazide derivatives 2 were condensed with CS2 under basic conditions (KOH in ethanol)25) to provide the 1,3,4-oxadiazole core structure 5 bearing the thiol functionality at the C-5 position. The benzimidazole pharmacophore was then conjugated into molecule through a methylthioether linker via nucleophilic substitution reaction with 2-(chloromethyl)benzimidazole derivatives 637) under mild basic conditions (sodium acetate) to afford the desired benzimidazole-1,3,4-oxadiazole hybrids (7a–s) in reasonable to good total yields (37–74%) (Table 1). Different substituents were introduced to the positions N-3″ and C-6″ of the benzimidazole ring to investigate their electronic effect on activities.
Table 1. Synthesis and Cytotoxicities of 1,3,4-Oxadiazole/Benzimidazole Hybrids
 |
|---|
| Compd. | R1 | R2 | R3 | Yielda)(%) | IC50 (µM) |
|---|
| HeLa | MCF-7 | A549 |
|---|
| 4a | 3,4,5-(OCH3)3 | H | H | 79 | 89.89 ± 1.70 | >100 | >100 |
| 4b | 3,4,5-(OCH3)3 | Bn | H | 83 | 44.81 ± 0.77 | 6.92 ± 1.38 | 38.34 ± 2.32 |
| 4c | 3,4,5-(OCH3)3 | 3-F-Bn | H | 78 | 30.81 ± 0.57 | >100 | >100 |
| 4d | 3,4,5-(OCH3)3 | 4-F-Bn | H | 85 | 3.23 ± 0.51 | 7.80 ± 0.55 | 5.96 ± 0.21 |
| 4e | 3,4,5-(OCH3)3 | 2-Cl-Bn | H | 87 | 37.54 ± 5.70 | 13.88 ± 0.73 | 58.79 ± 4.98 |
| 4f | 3,4,5-(OCH3)3 | 3,4-F2-Bn | H | 78 | >100 | >100 | >100 |
| 4g | 3,4,5-(OCH3)3 | CH2CH2OCH3 | H | 78 | >100 | 71.31 ± 6.12 | >100 |
| 4h | 3,4,5-(OCH3)3 | 3-Pyridinyl-CH2 | H | 84 | >100 | 33.38 ± 2.64 | >100 |
| 4i | 3,4,5-(OCH3)3 | 4-CH3-Bn | H | 86 | 3.59 ± 0.07 | 2.70 ± 0.30 | 6.64 ± 0.28 |
| 4j | 3,4-(OCH3)2 | H | H | 76 | >100 | >100 | >100 |
| 4k | 3,4-(OCH3)2 | Bn | H | 69 | >100 | >100 | >100 |
| 4l | 3,4-(OCH3)2 | 3-F-Bn | H | 69 | >100 | >100 | >100 |
| 4m | 3,4-(OCH3)2 | 2-Cl-Bn | H | 78 | >100 | >100 | >100 |
| 4n | 3,4-(OCH3)2 | 3,4-F2-Bn | H | 79 | >100 | >100 | >100 |
| 4o | 3,4-(OCH3)2 | CH2CH2OCH3 | H | 89 | >100 | >100 | >100 |
| 4p | 3,4-(OCH3)2 | 3-Pyridinyl-CH2 | H | 78 | >100 | >100 | >100 |
| 4q | 3,4-(OCH3)2 | 4-CH3-Bn | H | 85 | >100 | >100 | >100 |
| 7a38) | 3,4,5-(OCH3)3 | H | H | 65 | >100 | 82.55 ± 6.09 | >100 |
| 7b | 3,4,5-(OCH3)3 | Bn | H | 56 | 40.00 ± 1.49 | 19.51 ± 0.94 | 26.29 ± 2.24 |
| 7c | 3,4,5-(OCH3)3 | 4-F-Bn | H | 54 | 15.85 ± 0.44 | 20.43 ± 1.41 | 22.21 ± 0.77 |
| 7d | 3,4,5-(OCH3)3 | 3,4-F2-Bn | H | 55 | >100 | >100 | >100 |
| 7e | 3,4,5-(OCH3)3 | CH2CH2OCH3 | H | 62 | >100 | >100 | >100 |
| 7f | 3,4,5-(OCH3)3 | CH2CH2OH | H | 55 | >100 | >100 | >100 |
| 7g | 3,4,5-(OCH3)3 | 4-CH3-Bn | H | 63 | >100 | 45.59 ± 2.23 | >100 |
| 7h | 3,4,5-(OCH3)3 | Cyclohexyl | H | 62 | 22.27 ± 0.52 | 11.66 ± 0.71 | 37.05 ± 1.45 |
| 7i38) | 3,4,5-(OCH3)3 | H | NO2 | 57 | >100 | 21.75 ± 2.82 | >100 |
| 7j | 3,4,5-(OCH3)3 | H | Cl | 38 | >100 | 38.02 ± 2.24 | >100 |
| 7k | 3,4,5-(OCH3)3 | H | CH3 | 63 | 52.75 ± 1.63 | 51.17 ± 3.0 | 61.75 ± 3.07 |
| 7l | 3,4,5-(OCH3)3 | Bn | OCH3 | 62 | >100 | >100 | >100 |
| 7mb) | 3,4-(OCH3)2 | H | H | 74 | 94.52 ± 4.51 | 34.20 ± 0.58 | >100 |
| 7n | 3,4-(OCH3)2 | Bn | H | 54 | 56.87 ± 2.33 | 24.94 ± 0.98 | >100 |
| 7o | 3,4-(OCH3)2 | 2-Cl-Bn | H | 64 | >100 | >100 | >100 |
| 7p | 3,4-(OCH3)2 | 4-CH3-Bn | H | 69 | >100 | >100 | >100 |
| 7q | 4-OH | H | H | 55 | >100 | 66.15 ± 3.77 | 66.26 ± 1.28 |
| 7rb) | 3,4-(OCH3)2 | Bn | Cl | 62 | >100 | >100 | >100 |
| 7s | 3,4-(OCH3)2 | Bn | CH3 | 59 | 55.34 ± 3.56 | 26.35 ± 0.83 | >100 |
| 5-FUc) | — | — | — | 29.9 ± 1.63 | 35.4 ± 4.56 | 27.9 ± 3.90 |
a) Total isolated yield. b) Compounds are commercially available. c) 5-Fluorouracil (5-FU) was used as a positive control. Data are presented as mean ± standard deviation (S.D.) (n = 3); Bn: benzyl.
All of the synthesized 1,3,4-oxadiazole derivatives 4a–q and 7a–s were tested for their cytotoxicities against the three human cancer cell lines, including cervical cancer (HeLa), breast cancer (MCF7), and lung cancer (A549) cell lines. The results are summarized in Table 1. The assay indicated that compounds 4f, 4j–q, 7d–f, 7l, 7o–p, and 7r showed no activity at 100 µM against all tested cancer cell lines. Comparable to stronger activities with respect to the positive control 5-FU towards one cell line was found for compounds 4c (HeLa) and 4b, 4e, 4h, 7i–j, 7m–n, 7s (MCF-7), in which compound 4b (IC50 = 6.92 µM) illustrated almost 5-fold activity against the MCF-7 cell line compared to 5-FU (IC50 = 35.4 µM). Simultaneous cytotoxic effects consistent with 5-FU on two cancer cell lines were observed for compounds 7b (MCF-7, A549) and 7h (HeLa, MCF-7). Four compounds exhibited good cytotoxicities against the three tested cell lines (HeLa, MCF-7, A549) including 4d (IC50 = 3.23, 7.8, 5.96 µM), 4i (IC50 = 3.59, 2.7, 6.64 µM), 7c (IC50 = 15.85, 20.43, 22.21 µM) and 7h (IC50 = 22.7, 11.66, 37.05 µM), respectively. Notably, compounds 4d and 4i demonstrated 9- and 8-fold (HeLa), 4.5- and 13-fold (MCF-7), 4.7- and 4-fold (A549) higher activity compared to the positive control 5-FU [IC50 = 29.9 µM (HeLa), 35.4 µM (MCF-7) and 27.9 µM (A549)], respectively. The cytotoxicity of compound 4i (IC50 = 2.7 µM) was higher than that of the reported unsubsituted benzimidazole conjugated oxadiazole II19) (IC50 = 5.0 µM) or the thioether linked oxadiazole-metronidazole hybrid IV24) (IC50 = 10.6 µM) as shown in Fig. 2.
SAR AnalysisThe SAR analysis of the synthesized benzimidazole/oxadiazole hybrids (4a–q) and (7a–s) revealed that the cytotoxicity of these compounds depends essentially on the attached group (R2) at the position N-1 and (R3) at C-5 of the benzimidazole moiety with the benzyl or 3-pyridinylmethyl group at N-1 position showed better cytotoxicity than the one with aliphatic chain (4g, 4o, 7e, 7f). Furthermore, the presence of the methylthioether linkage combined with the 3,4,5-(OCH3)3-phenyl (Ph) moiety on one side of the oxadiazole ring seemed to be worse as there was a noticeable decrease in activity of the compound compared to its analogue without the linkage (comparing 7c with 4d, 7g with 4i, 7e with 4g, 7l with 4b). In contrast, the effect of the thioether linker was reverse for the 3,4-(OCH3)2-Ph analogues that induced a remarkable increase in cytotoxicity towards the MCF-7 cells (comparing 7m with 4j and 7n with 4k).
Within the series A (compounds 4a–q), all compounds with the 3,4-(OCH3)2-Ph moiety at C-2 of the oxadiazole ring (compounds 4j–q) or with the free N–H group (compound 4a) were inactive. For compounds bearing the 3,4,5-(OCH3)3-Ph at C-2 position, investigation of R2 moiety showed that the presence of the phenyl group in compound 4b resulted in increasing cytotoxicity, especially on the MCF-7 cell line which was more than 14-fold increase in activity compared to 4a with the free N-H moiety. Notably, the additional introduction of a hydrophobic group on the benzene ring such as F (4d) or CH3 (4i) seemed to be crucial leading to the remarkable increase in activity. Accordingly, compound 4d with the F group showed 14-fold and 6-fold higher activity against the HeLa and A549 cell lines, respectively, and remained the activity on the MCF-7 compared to 4b. Importantly, the CH3 group in compound 4i not only induced stronger activity against HeLa and A549 (12- and 6-fold, respectively) but also resulted to 2.5-fold increase activity against the MCF-7 cell lines compared to 4b. Noticeably, the introduction of one more F group at C-3 position (4f) just led to complete loss of the activity compared to 4d, propably due to the steric hindrance of the two F groups that subsequently prevent the interaction with the receptor. Beside the electronic characteristic, the positions of the substituent on the phenyl ring showed the varying degree of inhibition against the cancer cell lines. Comparison of the structures of 4c, 4e bearing the halogen group at the C-2, C-3 position, respectively, with compound 4d revealed that the best activity was obtained for the C-4 position. The same results were also observed in the case of the 3-pyridinyl substituent (compound 4h) although comparable activity to the control 5-FU against the MCF-7 cell line still remained. These findings for the series A suggest that the 3,4,5-(OCH3)3-Ph moiety at the C-2 position of the oxadiazole ring in combination with the hydrophobic (4-F, 4-CH3) groups at N-1 position of the benzimidazole ring contribute to anticancer activities. Without substituent on the benzene ring as in the case of 4b retains only its activity against MCF-7 with less active on other cancer cell lines.
Within the series B, the same critical effect of a lipophilic substituent such as 4-F-Bn (compound 7c), 4-CH3-Bn (7g), cyclohexyl group (7h) at the N-1 position on the activity was also observed. Noticeably, compound 7h bearing the cyclohexyl group induced 7-fold increase in activity against the MCF-7 compared to 7a with the free N–H position. Substituents at C-5 position of the benzimidazole ring also play an important role. As the results, strong electron withdrawing groups such as NO2 (7i) or Cl (7j) enhanced 4- or 2-fold actitivity compared to 7a, respectively, against the MCF-7 cells. In contrast, strong electron donating group like OCH3 just led to complete loss of the activity. An inverse effect was observed for the 3,4-(OCH3)2-Ph analogs with which the presence of Cl group (compound 7r) led to entire loss of the acitivity towards the HeLa and MCF-7 compared to 7n, while the CH3 group (compound 7s) still helped remain the cytotoxicity against the MCF-7 cell line. Finally, the 4-OH-Ph group at C-2 of the oxadiazole (compound 7q) appeared not to play the role for the cytotoxicity towards the tested cancer cell lines.
Figure 3 summarizes crucial structural features of the potent cytotoxic benzimidazole conjugated 1,3,4-oxadiazoles in which compound 4b (R2 = Bn) was found as lead compounds against the MCF-7 while compounds 4d (R2 = 4-F-Bn) and 4i (R2 = 4-CH3-Bn) as leads towards all three tested cancer cell lines. Previous studies have indicated that compounds IIIa and IIIb (Fig. 1) possessed potent inhibitory activtities against thymidylate synthase (TS), which is the important target for cancer treatment to prevent the cell growth and proliferation.23) Thus, further investigation of the inhibitory effect of the derivatives, especially compound 4i, on the TS activity may present an excellent platform for developing benzimidazole-conjugated 1,3,4-oxadiazoles-based antitumor agents.
Conclusion
We have reported the design and synthesis of thirty-two novel benzimidazole conjugated 1,3,4-oxadiazoles and their in vitro cytotoxicities against the HeLa, MCF7, and A549 human cancer cell lines. As the results, compounds 4b, 4e and 7h exhibited good activities against the MCF-7, in which the lead compound 4b possessed 5-fold activity compared to 5-FU. Furthermore, compounds 4d and 4i were found as leads towards all three tested cancer cell lines which exhibited 4.5–9-, 4.2–8-fold higher activity compared to the control 5-FU, respectively. These results demonstrate that the benzimidazole/1,3,4-oxadiazole hybrid represents potent structural hybrid motifs that might contribute to the development of structurally interesting anti-cancer agents.
Experimental
ChemistryGeneral InformationReactions were monitored by TLC on 0.2 mm pre-coated silica-gel 60 F254 plates (Merck). 1H- and 13C-NMR spectra were measured with Bruker Avance 500 and 600 MHz spectrometers. High resolution electrospray ionization (HRESI)-MS observations were performed on a Sciex OS 1.2 mass spectrometer. Fourier transform (FT)-IR was conducted using the KBr pellet method on a Thermo Nicolet 6700 spectrometer. Chemical shifts are given in parts per million (ppm) relative to tetramethylsilane (Me4Si, δ = 0); J values are given in Hertz. All chemicals and solvents used in this study were of analytical grade.
General Procedure for the Synthesis of Benzimidazole Conjugated Oxadiazole Derivatives (4a–q)A mixture of the benzohydrazide 2 (0.2 mmol) and aldehydes 3 (1 equiv.) in absolute ethanol (4 mL) was stirred at 80 °C for 4–9 h. After cooling down to room temperature, the excess solvent was evaporated under reduced pressure to obtain the intermediate imines which were used for the next step without further purification.
The obtained intermediate imines (0.2 mmol), I2 (3 equiv.) and K2CO3 (5 equiv.) were dissolved in DMSO (1 mL). The reaction mixture was stirred at 80 °C for 4–8 h. After completion of the reaction, water (40 mL) was added and the excess amount of I2 was quenched by aqueous saturated solution of Na2S2O3. The resulting mixture was extracted with ethyl acetate (3 × 25 mL). The combined organic layers were washed with water, dried over anhydrous Na2SO4 and solvent was evaporated. Purification of the crude products by silica gel column chromatography afforded the desired 1,3,4-oxadiazole derivatives (4a–q).
General Procedure for the Synthesis of Thioether Linked 1,3,4-oxadiazole/benzimidazole Hybrids (7a–s)A mixture of 5 (0.3 mmol), benzimidazoles 637) (1.2 equiv.), and NaOAc (3 equiv.) in DMF (0.5 mL) was stirred with at 80 °C for 15 min. After evaporation of the sovent under reduced pressure, the residue was dissolved in ethyl acetate (20 mL) and the organic layer was washed with water (3 × 40 mL), dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography to give the deisred 1,3,4-oxadiazole/benzimidazole derivatives (7a–s).
Cytotoxicity EvaluationSample PreparationAll synthesized compounds were dissolved in DMSO to make 10 mM stock solutions. Serial dilutions were prepared in culture medium. The positive control, 5-FU, was dissolved in DMSO to make a 10 mM stock solution and then stored at −20 °C until use.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium Bromide (MTT) Proliferation AssayThe cytotoxic activities of the synthesized compounds were evaluated against human cancer cell lines (HeLa, MCF-7, and A549), using the MTT assay with some modifications.39) The human cancer cell lines were cultured in α-minimum essential medium (α-MEM), supplemented with 1% antibiotic antimycotic solution and 10% fetal bovine serum, at 37 °C and in a 5% CO2 atmosphere. Cells at 80–90% confluence were harvested and centrifuged at 3000 rpm for 3 min. The supernatant was discarded and the cell pellet was resuspended in fresh medium. Aliquots (100 µL) of the cells were seeded in 96-well plates (1 × 104 cells/well) and incubated for 24 h. The cells were then washed with phosphate-buffered saline (PBS), and various concentrations of tested compounds, including the positive control, 5-FU (5–100 µM), were added to the wells. After a 72 h incubation, the cells were washed with PBS, and 100 µL aliquots of medium containing MTT solution (5 mg/mL) were added to each well and incubated for 3 h. The absorbance was recorded using a microplate reader at 570 nm. Percent proliferation inhibition was calculated using the following formula:
-
At: Absorbance of test compound, Ab: Absorbance of blank, Ac: Absorbance of control.
The concentrations (IC50 values) of the compounds required to inhibit 50% of the growth of the human cancer cell lines were calculated based on the relationship between concentrations and percent inhibitions, using the GraphPad Prism 5.0 software. Each experiment was performed three times, and all data are presented as mean ± standard deviation (S.D.).
Acknowledgments
This research was funded by Vietnam Ministry of Education and Training (Grant number: B2020-TCT-12).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
References
- 1) Morishita K., Ito Y., Otake K., Takahashi K., Yamamoto M., Kitao T., Ozawa S., Hirono S., Shirahase H., Chem. Pharm. Bull., 69, 333–351 (2021).
- 2) Huang M., Ren J., Wang Y., Chen X., Yang J., Tang T., Yang Z., Li X., Ji M., Cai J., Chem. Pharm. Bull., 69, 620–629 (2021).
- 3) Nakka N., Munnisa B., Duvvala P., Kalam S., Porika M., Chandrashekar C., Vedula R. R., Vanga M. R., Garlapati A., Chem. Pharm. Bull., 68, 1170–1177 (2020).
- 4) Liu W., Wang G., Peng Z., Li Y., Chem. Pharm. Bull., 68, 1184–1192 (2020).
- 5) Saini M. S., Kumar A., Dwivedi J., Singh R., Intl. J. Pharm. Sci. Res., 4, 66–77 (2013).
- 6) Verma G., Khan M. F., Akhtar W., Alam M. M., Akhter M., Shaquiquzzaman M., Mini Rev. Med. Chem., 19, 477–509 (2019).
- 7) Salahuddin M. A., Yar M. S., Mazumder R., Chakraborthy G. S., Ahsan M. J., Rahman M. U., Synth. Commun., 47, 1805–1847 (2017).
- 8) Kakkar S., Narasimhan B., BMC Chemistry, 13, 1–24 (2019).
- 9) Kumar K. A., Jayaroopa P., Kumar G. V., Intl. J. ChemTech. Res., 7, 1782–1791 (2012).
- 10) Sharma S., Sharma P. K., Kumar N., Dudhe R., Der. Pharm. Chemica., 2, 253–263 (2010).
- 11) Gaba M., Mohan C., Med. Chem. Res., 25, 173–210 (2016).
- 12) Guimaraes C. R. W., Boger D. L., Jorgensen W. L., J. Am. Chem. Soc., 127, 17377–17384 (2005).
- 13) Nayak S., Gaonkar S. L., Musad E. A., Dawsar A. M. A., J. Saudi Chem. Soc., 25, 101284 (2021).
- 14) Yoon Y. K., Ali M. A., Wei A. C., Shirazi A. N., Parang K., Choon T. S., Eur. J. Med. Chem., 83, 448–454 (2014).
- 15) Yoon Y. K., Ali M. A., Wei A. C., Choon T. S., Osman H., Parang K., Shirazi A. N., Bioorg. Med. Chem., 22, 703–710 (2014).
- 16) Shrivastava N., Naim M. J., Alam M. J., Nawaz F., Ahmed S., Alam O., Arch. Pharm., 350, e201700040 (2017).
- 17) Baig M. F., Nayak V. L., Budaganaboyina P., Mullagiri K., Sunkari S., Gour J., Kamal A., Bioorg. Chem., 77, 515–526 (2018).
- 18) Husain A., Rashid M., Mishra R., Parveen S., Shin D. S., Kumar D., Bioorg. Med. Chem. Lett., 22, 5438–5444 (2012).
- 19) Akhtar M. J., Siddiqui A. A., Khan A. A., Ali Z., Dewangan R. P., Pasha S., Yar M. S., Eur. J. Med. Chem., 126, 853–869 (2017).
- 20) Yurttaş L., Demirayak Ş., Çiftç G. A., Yıldırım Ş. U., Kaplancıklı Z. A., Arch. Pharm. Chem. Life Sci., 346, 403–414 (2013).
- 21) Çevik U. A., Osmaniye D., Çavuşoğlu B. K., Sağlik B. N., Levent S., Ilgin S., Can N. Ö., Özkay Y., Kaplancikli Z. A., Med. Chem. Res., 28, 2252–2261 (2019).
- 22) Abd El-Meguid E. A., Awad H. M., Anwar M. M., Russ. J. Gen. Chem., 89, 348–356 (2019).
- 23) Alam M. M., Almalki A. S., Neamatallah T., Ali N. M., Malebari A. M., Nazreen S., Pharmaceuticals, 13, 390 (2020).
- 24) Du Q. R., Li D. D., Pi Y. Z., Li J. R., Sun J., Fang F., Zhong W. Q., Gong H. B., Zhu H. L., Bioorg. Med. Chem., 21, 2286–2297 (2013).
- 25) Mochona B., Mazzio E., Gangapurum M., Mateeva N., Redda K. K., Chem. Sci. Trans., 4, 534–540 (2015).
- 26) Bansal Y., Silakari O., Bioorg. Med. Chem., 20, 6208–6236 (2012).
- 27) Hue T. B. B., Quy T. K., Won K. O., Duy D. V., Yen N. T. C., Cuc T. K. T., Em C. P., Phuong T. T., Loan T. T., Hieu V. M., Tetrahedron Lett., 57, 887–891 (2016).
- 28) Hue B. T. B., Nguyen H. M., Hieu M. V., Thanh D. L. D., Son N. H., De T. Q., Morita H., Heterocycles, 98, 650–665 (2019).
- 29) Hue B. T. B., Kiep M. D., Huy T. D. N., Hieu V. M., Thanh L. D. D., De Q. T., Morita H., Tetrahedron, 98, 132426 (2021).
- 30) Shawky A. M., Ibrahim N. A., Abdalla A. N., Abourehab M. A., Gouda A. M., J. Enzyme Inhib. Med. Chem., 36, 1313–1333 (2021).
- 31) Abolhasani A., Heidari F., Noori S., Mousavi S., Abolhasani H., Curr. Chem. Biol., 14, 38–47 (2020).
- 32) Polothi R., Raolji G. S. B., Kuchibhotla V. S., Sheelam K., Tuniki B., Thodupunuri P., Synth. Commun., 49, 1603–1612 (2019).
- 33) Labruere R., Helissey P., Desbene-Finck S., Giorgi-Renault S., Lett. Org. Chem., 9, 568–571 (2012).
- 34) Hue B. T. B., Nguyen H. P., De T. Q., Hieu V. M., Jo E., Tuan N. V., Thoa T. T., Anh L. D., Son N. H., Thanh D. L. D., Dupont-Rouzeyrol M., Grailhe R., Windisch M. P., ChemMedChem, 15, 1453–1463 (2020).
- 35) Jiang Z. Q., Miao D. Z., Tong Y., Pan Q., Li X. T., Hu R. H., Han S. Q., Synthesis, 47, 1913–1921 (2015).
- 36) Niu P., Kang J., Tian X., Song L., Liu H., Wu J., Yu W., Chang J., J. Org. Chem., 80, 1018–1024 (2015).
- 37) Bai Y. B., Zhang A. L., Tang J. J., Gao J. M., J. Agric. Food Chem., 61, 2789–2795 (2013).
- 38) Sule D. P., Shah M. H., Ghooi S. R., Bhide M. B., Bull. Haffkine, 7, 17–21 (1979).
- 39) Mosmann T., J. Immunol. Methods, 65, 55–63 (1983).