Reviews
An Update Mini-Review on the Progress of Azanucleoside Analogues
2022 年 70 巻 7 号 p. 469-476

詳細
2022 年 70 巻 7 号 p. 469-476
The development of structurally novel nucleoside analogues is an active area in medicinal chemistry, since these drugs have proven clinical efficacy for decades. Azanucleosides are nucleoside analogues in which the sugar moieties are composed of nitrogen-containing rings or chains. In recent years, many azanucleosides have demonstrated therapeutic potential. In this short review, we describe recent advancements in azanucleosides, which may translate in a better understanding of the molecular design, biological activity, structure–activity relationship, and their related mechanism of action. The information summarized in this paper should encourage medicinal chemists in their future efforts to create more potent and effective chemotherapeutic agents.
Nucleosides are endogenous compounds that not only serve as building blocks for DNA and RNA synthesis, but also involve in various biological processes, including cell signaling, enzyme regulation, and metabolism. Since natural nucleosides play an essential role in cell function, synthesizing their structurally similar analogues as chemotherapeutic agents have proven effective.1–4) Nucleoside analogues now account for the major part of antimetabolites. The clinical use of more than 30 nucleoside analogues have been approved for viral infections and cancers.5–7) The U. S. Food and Drug Administration (FDA) has approved remdesivir and molnupiravir for corona virus disease 2019 (COVID-19), reminding us of nucleoside analogues’ pivotal role in medicine.8,9) The design and synthesis of nucleoside analogues to combat various diseases remain an exciting drug development area.
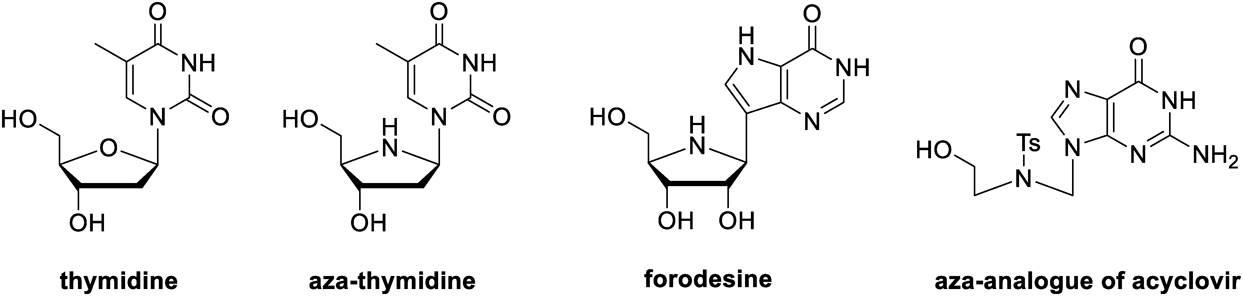
In general, novel analogues of nucleosides can be synthesized by modifying the heterocyclic base, the sugar moiety, and the glycosidic bond of the nucleoside scaffold. Among the diverse analogues, azanucleosides are sugar-modified analogues characterized by substituting a nitrogen atom for the sugar endocyclic oxygen atom.10,11) Nitrogen can act as an additional hydrogen bond donor, and its heteroatom effect can increase the interaction of nucleoside analogues with biological targets.6,12) Azanucleosides can be divided into two types based on the linkage between the sugar moiety and the base moiety, namely C-azanucleosides and N-azanucleosides. The former contains the glycosidic C–N bond, like aza-thymidine (Fig. 1), while the latter contains the glycosidic C–C bond, exemplified by forodesine. It is worth mentioning that the endocyclic nitrogen atoms of N-azanucleosides are conveniently protected since the N,N-acetals in N-azanucleosides, such as aza-thymidine (Fig. 1), are prone to spontaneous hydrolysis.11,13–19) However, glycosidic C–C bonds are resistant to acid-catalyzed and enzymatic hydrolysis, making C-azanucleosides more stable.20) In some cases, azaribofuranosyl moieties are replaced by chains containing nitrogen atoms, and these molecules are termed acyclic azanucleosides, such as aza-analogue of acyclovir (Fig. 1). The inherent flexibility of acyclic nucleosides may translate in an enhanced interaction with the target binding sites in some cases.
To date, azanucleoside analogues are increasingly attractive because they exhibit a variety of pharmacological activities, such as anticancer, antiviral, and antimicrobial properties. Hernández and Boto’s work in 2014 is known to be the latest review of azanucleoside derivatives.10) This mini-review updates the recent advancements of azanucleosides. In this article, we highlight the biological activities of azanucleosides and hope to provide valuable information for developing new nucleoside analogues as potential drug candidates.
The current review examines the recent development of azanucleoside analogues as potential chemotherapeutic agents. This article has compiled and presented a collection of information regarding drug design strategy, biological evaluation, structure–activity relationship, and mechanism of action.
2.1. Inhibitors of Purine Nucleoside Phosphorylases (PNPs)Purine nucleoside phosphorylases (PNPs) are pivotal purine metabolizing enzymes responsible for converting purine nucleosides into ribose or deoxyribose 1-phosphate and purine bases via phosphorolysis (Fig. 2A). Individuals with genetically deficient PNP have impaired T-cell function. Due to the absence of PNP, phosphorolysis of deoxyguanosine (dGuo) is arrested in T-cells, causing deoxyguanosine triphosphate (dGTP) to accumulate. In high concentrations of dGTP, ribonucleotide reductase is inhibited, which disrupts deoxynucleotide pool balance, resulting in apoptosis in T-cells. As a result of this discovery, the rational design of human PNP (hPNP) inhibitors can be applied to the treatment of T-cell lymphomas and autoimmune disorders.21,22)
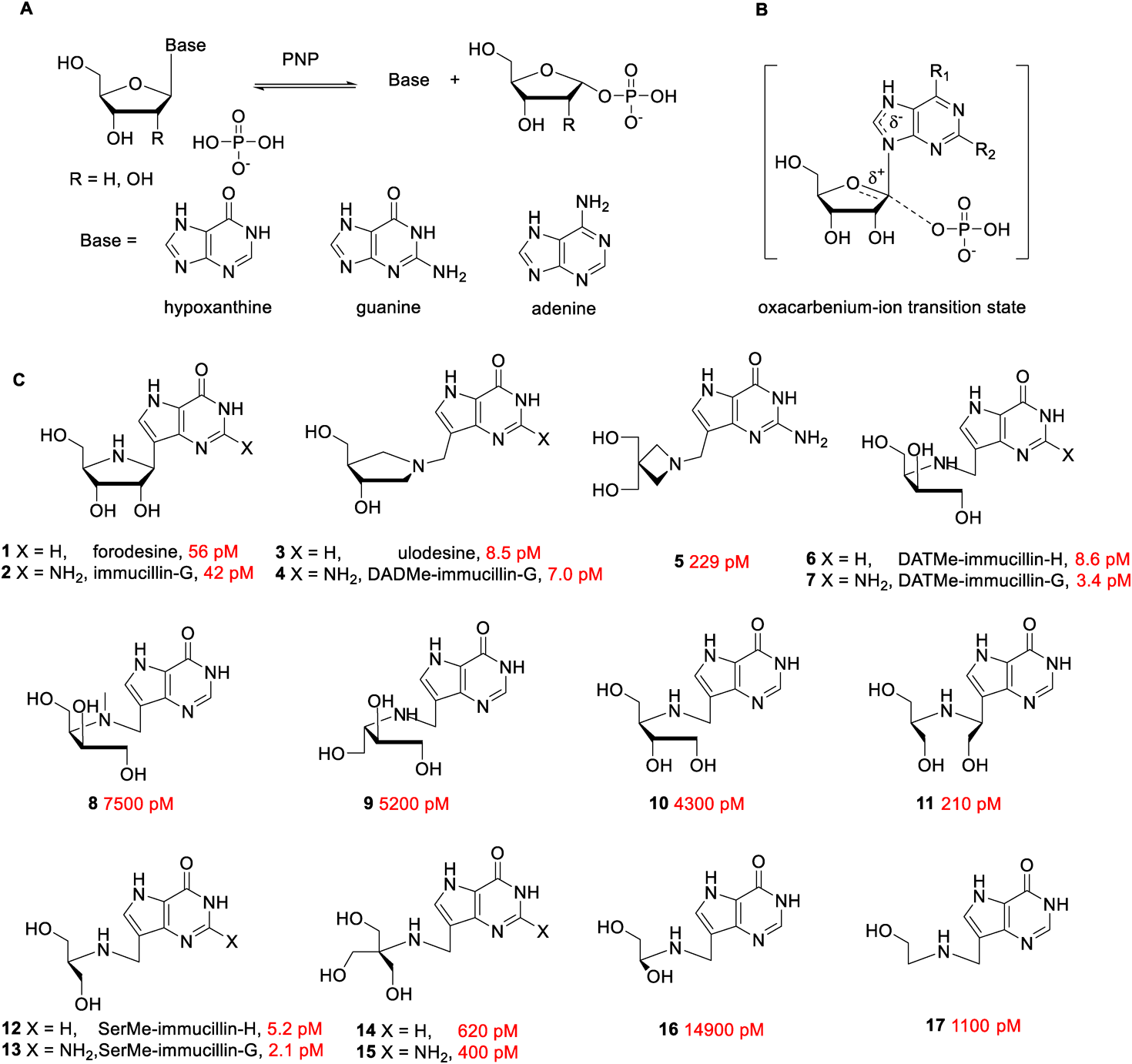
The dissociation constants (Ki) for human purine nucleoside phosphorylase (hPNP) are colored red.
Using the kinetic isotope effect (KIE) and computational quantum chemistry, Evans et al. proposed the mechanism catalyzed by PNP and elucidated the process of phosphorolysis through a nucleophilic substitution reaction. In the enzymatic active site of PNP, the phosphate nucleophile attacked the anomeric carbon with the formation of oxocarbenium-ions (Fig. 2B), which further decomposed into nucleobase and sugar 1-phosphate.23) The structured character of the enzymatic transition state provided a blueprint for the novel PNP inhibitor design. Forodesine 1 (BCX1777, immucillin-H) and immucillin-G 2 were typical first-generation transition state analogues inhibitors for PNP, composed of aza-ribose and purine bases through a C–C bond (Fig. 2C). Under physiological conditions, the imino group easily get protonated, conferring an azasugar moiety with a ribocation feature, and C–C glycosidic bond stabilized the structure to match the early dissociative transition state. The dissociation constants (Ki) of forodesine and immucillin-G for hPNP were found to be 56 and 42 pM, respectively.24) The second-generation transition state analogues inhibitors, exemplified by ulodesine 3 (BCX4208, DADMe-immucillin-H) and DADMe-immucillin-G 4 (BCX4945), were designed based on the late transition state, in which purine leaving groups were fully dissociated, and cationic centers tended to form at anomeric sites. To better capture the characteristic of the late transition state, methylene was used to bridge the purine base and the nitrogen atom in the pyrrolidine ring component. Compared to first-generation enzyme inhibitors, ulodesine 3 and DADMe-immucillin-G 4 exhibited a more potent inhibition activity, whose dissociation constants were reported to be 8.5 and 7.0 pM, respectively.25) Compound 5, an azetidine-based derivative of DADMe-immucillin-G, was a 229 pM hPNP inhibitor, indicating that the structure of azasugar affects enzymatic inhibition activity. Various structurally diverse acyclic azasugars were adopted to construct next-generation transition state analogue inhibitors, such as compounds 6–17. As depicted in Fig. 2C, DATMe-immucillin-G 7 (Ki = 3.4 pM) and SerMe-immucillin-G 13 (Ki = 2.1 pM) displayed the lowest dissociation constants never before, wherever other acyclic aza-analogues exerted less efficacy.26) The cocrystal structures of hPNP with some typical transition-state analogue inhibitors (1, 3, 6, and 12) indicated that these molecules could bind tightly with the active site.27) Among the above-mentioned hPNP inhibitors, forodesine 1 underwent successful clinical trials in Japan and was approved for treating relapsed/refractory peripheral T- cell lymphoma (PTCL) in 2017.28) Ulodesine 3 has completed phase II clinical trials for the treatment of gout to date. As a severe auto-inflammatory disease, gout is associated with a high uric acid level, produced by purine oxidation. Excess uric acid could be efficiently controlled by limiting the formation of purine precursors by inhibiting hPNP.29)
In addition to utility in treating T-cell cancers and gout, PNP inhibitors have potential application against protozoan parasites. As the most lethal causative agent for human malaria, Plasmodium falciparum (Pf) is a purine auxotroph and depends primarily upon hypoxanthine produced by PNP via the purine salvage pathway for purine nucleotide synthesis. It has been established that some hPNP inhibitors in Fig. 2C could inhibit PfPNP while generally displaying less efficacy. Forodesine 1 was an 860 pM inhibitor for PfPNP, approximately 15 times less potent for hPNP. The Plasmodium falciparum living inside the erythrocytes could be killed by the addition of forodesine 1.30) With a dissociation constant of 7 pM for hPNP and 890 pM for PfPNP, DADMe-immucillin-G 4 had been tested in the Aotus monkey model as a potential antimalarial agent.31) Infected Aotus monkeys were treated with 50 mg/kg of DADMe-immucillin-G 4 twice a day for seven days, which not only resulted in reduction in blood Plasmodium falciparum count (below 10−1 parasites/µL), but additionally no drug-related toxicity was detected. However, the parasites in treated animals began to regrow, following termination of therapy.
2.2. Inhibitors of Viral RNA-Dependent RNA Polymerase (RdRp)Galidesivir 18 (BCX4430, immucillin-A) was initially developed as a novel anti-trichomoniasis drug. As the causative agent for trichomoniasis, Trichomonas vaginalis is purine auxotrophic type and expresses rare PNP (TvPNP) in the purine salvage pathway. Compared to the above-mentioned hPNP or pfPNP, TvPNP tends to adopt adenosine as the substrate, and substrate specificity offers the opportunity to design antiparasitic drugs.32) Galidesivir 18 is an inhibitor of 87 pM for TvPNP. Nevertheless, its anti-trichomoniasis activity was not as expected against Trichomonas vaginalis even at 10 µL dosage of galidesivir 18. The loss of anti-trichomoniasis activity was due to the presence of some other purine salvage enzymes, and TvPNP did not play an essential role in the supply of purines.33) However, galidesivir 18 displayed remarkably broad-spectrum antiviral activity in the antiviral tests. More than 20 viral species in ten different virus families were found to be inhibited by galidesivir in vitro, such as filoviruses, togaviruses, bunyaviruses, arenaviruses, flaviviruses, and phenuiviruses, where a significant and strong antiviral activity was observed against filovirus and flavivirus induced infections34–38) (Fig. 3). These two virus families contained numerous pathogenic and zoonotic viruses, such as the Ebola virus (EBOV), Marburg virus (MARV), Sudan virus (SUDV), West Nile virus (WNV), tick-borne encephalitis virus (TBEV), Kyasanur forest disease virus (KFDV), Zika virus (ZIKV), and yellow fever virus (YFV). Until now, no effective therapies are available for the above-mentioned viral infections. To the best of our knowledge, galidesivir 18 was the first chemical entity to demonstrate promising anti-filovirus and anti-flavivirus activity. Comprehensive antiviral results (EC50 values ranging from 2.33 to 14.1 µM) are summarized in Fig. 3.
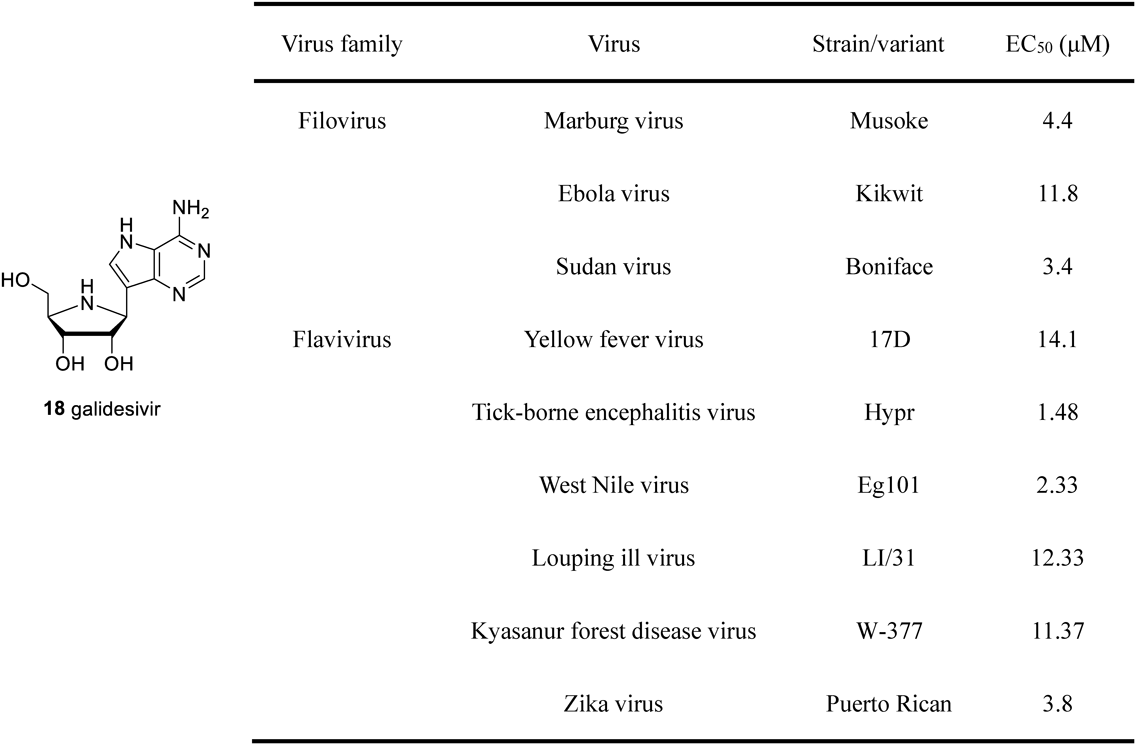
As an adenosine analogue, galidesivir 18 could be transformed into its triphosphate form through stepwise phosphorylation catalyzed by cellular kinases to resemble natural adenosine triphosphate. The triphosphate form is an inhibitor of viral RNA-dependent RNA polymerase (RdRp). The incorporated galidesivir triphosphate prevented further elongation of the RNA chain, thus reducing viral replication.34,38) The latest phase I clinical trial data confirmed good safety and tolerability of galidesivir 18.39) Besides, galidesivir 18 was pointed to be a potential therapeutic option for COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).40) Due to the fact that previous studies demonstrated galidesivir 18 potent antiviral activity against severe acute respiratory syndrome (SARS) and middle east respiratory syndrome coronavirus (MERS-CoV) of the coronaviridae family with EC50 values of 57.5 and 68.4 µM, respectively.34) A recent molecular docking study confirmed that galidesivir 18 could tightly bind to the RdRp of SARS-CoV-2 stain.41) The latest research indicated that galidesivir was potentially effective in treating novel coronavirus infections.42) The in vitro antiviral activity of galidesivir against SARS-CoV-2 in Caco-2 and Vero-76 cell line was evaluated by viral yield reduction (VYR) assay, which demonstrated EC90 values of 14.19 and 10.94 µM, respectively. In the hamster model of SARS-CoV-2 infection, early treatment with galidesivir (100 mg/kg twice a day) was found to reduce animals’ viral burden and body weight. Histopathological analysis indicated that the administration of galidesivir significantly reduced the damage to lung tissue and afforded a protective effect. The clinical efficacy of galidesivir has not yet been reported and needs to be characterized further.
2.3. Inhibitors of Human Thymidine Phosphorylase (hTP)Human thymidine phosphorylase (hTP) catalyzes thymidine to produce thymine and 2-deoxy-α-D-ribose-1-phosphate in the presence of phosphate (Fig. 4A). As a pivotal metabolic enzyme, hTP participates in the pyrimidine salvage pathway to guarantee cellular DNA’s reproductive and repaired progress. The high expression of hTP is associated with the pathogenesis of cancer by stimulating angiogenesis and antiapoptotic signaling. Therefore, hTP has been validated as an anticancer target.43) Inhibitors of hTP could be applied in anticancer therapy through antiangiogenic functions. A canonical example of this therapeutic strategy is tipiracil (TPI, 19). Coadministration of a thymidine phosphorylase inhibitor TPI can effectively resist the degradation of trifluorothymidine (TFT) and hence a chemically stable TFT demonstrates potent anticancer activity via the induction of DNA instability and double-stranded breaks. The chemotherapeutic agent containing a combination of trifluorothymidine and tipiracil had been approved by FDA as lonsurf for the treatment of metastatic colorectal cancer in 2015.44)
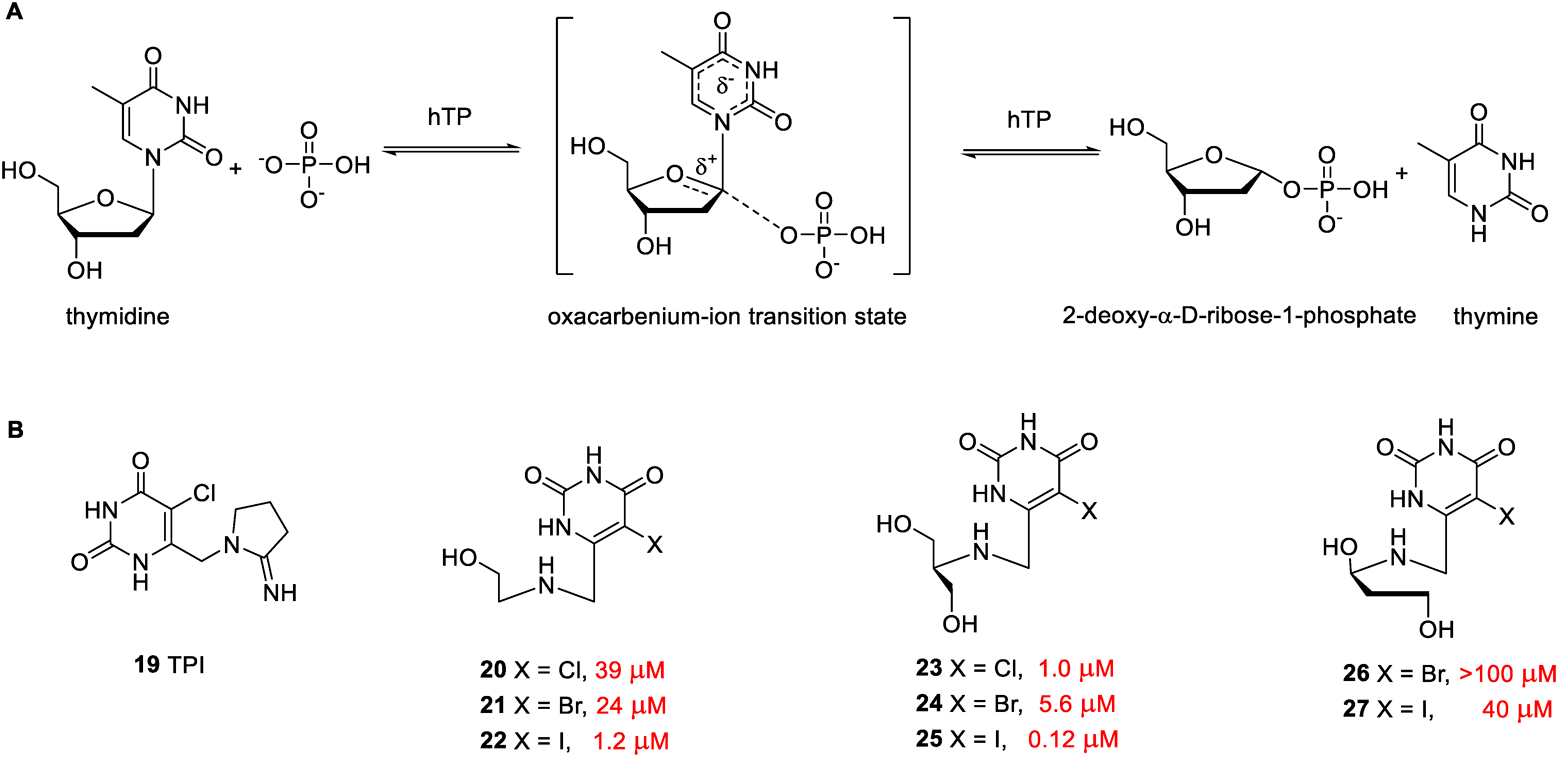
The inhibitory activities of compounds for human thymidine phosphorylase (hTP) are shown colored red.
Recently, Sperotto et al. reported some novel pyrimidine acyclic azanucleosides (compounds 20–27 in Fig. 4B) as human thymidine phosphorylase inhibitors.45) The structures of these hTP inhibitors were designed and inspired by an enzyme-catalyzed transition state (Fig. 4A). The introduction of halogen substituent polarized the heterocyclic ring moiety and stabilized the negative charge on the base part. At physiological pH, the nitrogen-containing acyclic sugar chain can be positively charged to mimic the oxocarbenium character of a transition state. Various acyclic sugar chains were attached to different halogenated pyrimidine rings to assess the relative potency of compounds against hTP activity. Most compounds in the series exhibited potent enzyme inhibition activity with EC50 values ranging from 0.12 to 40 µM, except compound 26 (EC50 > 100 µM). The enzyme screening results indicated that the electronegativity of halogens on the uracil ring affected the activity. Compared to chlorine and bromine, iodine atoms located at the 5-position of the heterocyclic ring can enhance enzyme inhibition activity. A second hydroxyl group was inserted at the β position, considering that nitrogen from the acyclic sugar chain, exerted improved activity such as compounds 23–25. The most active inhibitor of hTP was found to be compound 25, with an EC50 value of 0.12 µM. It is noteworthy that compound 25 inhibited hTP with a noncompetitive inhibition model, which cannot be reversed by either thymidine or phosphate substrates. The mechanism of hTP-inhibition was different from approved competitive inhibitor TPI. Compound 25 also displayed low cytotoxicity and genotoxicity. The administration of compound 25 at dosage of 50 mg/kg day for two weeks was found to significantly reduce tumor volume growth. Compound 25 in vivo anticancer activity was comparable to a first-line drug temozolomide, for treating glioblastoma multiforme. Immunohistochemistry (IHC) and histological analysis supported the capacity of this active compound to reduce vascular growth. Hence, it is apprehended that compound 25 could possibly be the most potent hTP inhibitor with the non-competitive inhibition model so far.
2.4. Inhibitors of the Heat Shock Response (HSR) PathwayThe HSR is an evolutionarily conserved cellular pathway that relieves various cellular stress and maintains protein homeostasis. When cells are exposed to stressful or harmful conditions such as extreme temperature, oxidative stress, heavy metals, or other cytotoxic chemical agents, cellular heat shock factors (HSFs) will be activated. As transcription factors, heat shock factors induce related genes to encode heat shock proteins (HSPs). The latter works as a molecular chaperone to facilitate protein’s correct folding or the removal of misfolded proteins to restore protein homeostasis.46,47) Cancer cells are characterized by overexpressed heat shock proteins owing to unique tumor microenvironment. A high level of heat shock proteins indicates a close relationship between carcinogenesis and drug resistance. Targeting the heat shock response pathway may thus lead to the discovery of novel anticancer agents.48,49)
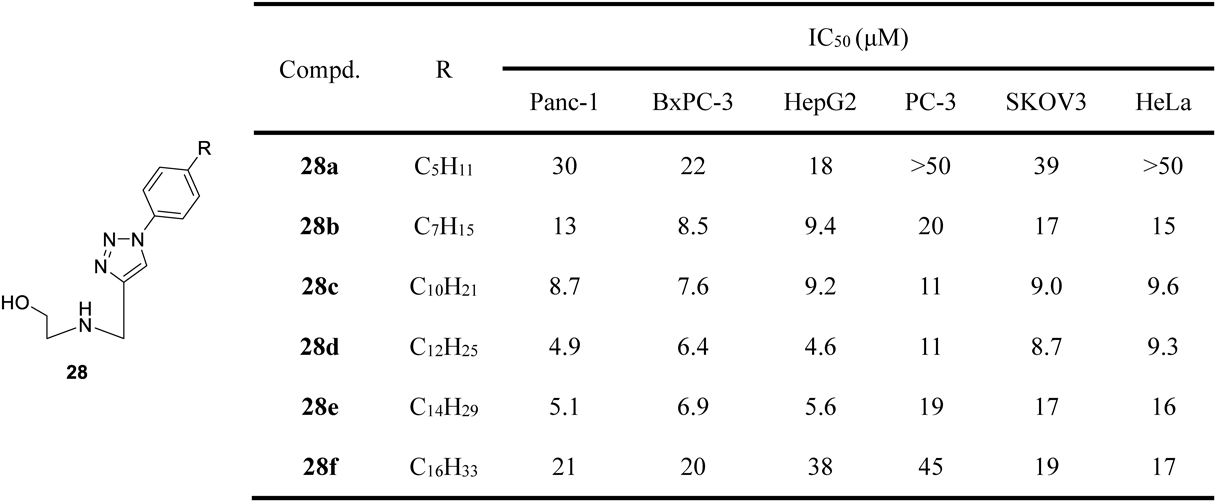
From 2009 to 2015, Peng and colleagues discovered that triazole nucleosides with expanded aryl moiety in the nucleobase showed potent anticancer activity and novel mechanisms of action, such as inhibition of heat shock factor 1 (HSF1), heat shock proteins, and related heat shock response pathway.50–52) Based on this finding, aryltriazole nucleosides may serve as promising anticancer lead compounds.53) Our recent work suggested that introducing nitrogen-containing groups at the terminal position of the acyclic sugar chain endowed aryltriazole nucleosides with improved physicochemical properties and better anticancer activity. Similarly, the amine-functioned aryltriazole nucleosides also inhibited cancer cell growth by targeting the heat shock response pathway.54) Based on these results, we developed novel aryltriazole acyclic C-azanucleosides 28 (Fig. 5) through our continuing drug discovery program.55) The series of acyclic azanucleosides were easily prepared via Huisgen 1,3-dipolar cycloaddition because their nucleobases were composed of aryl 1,2,3-triazole fragments. The results of the antiproliferative activity screening indicated that the substituents located in the aryl motif pronouncedly affected anticancer activity. Azanucleosides containing more than C3 hydrophobic alkyl chain in the aryl group (compounds 28a–f) showed potent antiproliferative activity against cancer cells with IC50 values below 50 µM. An appropriate alkyl chain (C12 alkyl chain) conferred acyclic azanucleoside 28d with the most potent antiproliferative activity with IC50 values ranging from 4.6 to 11 µM (Fig. 5). According to the results of the tests, modulating the hydrophilic-lipophilic balance may be an approach to obtain the desired molecules. The most active compound 28d was able to down-regulate the expression of HSF1, HSPs and their related client proteins, such as eukaryotic translation initiation factor 4E (eI4E), which was the client oncoprotein of HSP27. Hence, compound 28d is deemed appropriate to induce apoptosis by targeting the heat shock response pathway. We believe that these could be the most novel reported results depicting an acyclic azanucleoside showing such an anticancer mode of action and hence, may serve as a promising anticancer candidate.
Hypoxanthine–guanine–xanthine phosphoribosyltransferase (HGXPRT) of malarial parasites Plasmodium falciparum (Pf) is responsible for transferring the phosphoribosyl group from 5-phospho-α-D-ribosyl-1-pyrophosphate (PRPP) to 6-oxopurine bases with the formation of inorganic pyrophosphate (PPi) and nucleoside monophosphates (Fig. 6A). As nucleoside monophosphates are located in the central part of nucleotide biosynthesis, PfHGXPRT is proposed as a potential target in antimalarial chemotherapy.56) On the journey to discover new inhibitors of PfHGXPRT, some azanucleosides showed promising bioactivity. Schramm and colleagues designed the first-generation PfHGXPRT inhibitor based on transition state theory, exemplified by immucillinHP 2957) (Fig. 6B). The azasugar moiety of immucillinHP 29 well mimicked the ribocation feature of the transition state. This transition state analogue exhibited a powerful affinity for PfHGXPRT, with dissociation constants as low as 1.0 nM. However, the exposed phosphate group of immucillinHP 29 was vulnerable in the presence of phosphomonoesterases. To circumvent this labile structure, a more stable phosphonate group was introduced. Meanwhile, flexibly acyclic amino alcohols were brought to create next-generation PfHGXPRT inhibitors, such as compounds 30 and 31. The second-generation inhibitors 30 and 31 were characterized by simplified structures but maintained potent enzyme inhibition activity with EC50 values of 0.65 and 10.6 nM, respectively. However, the antimalarial activity of these phosphonate inhibitors against cultured parasites was disappointing. The existence of free phosphonate groups made these molecules too polar to cross the cellular membrane. To increase cellular permeability, the phosphonate group was masked with long alkyl chains to obtain the lysophospholipid prodrugs 32a–d. Most lysophospholipid prodrugs in the class could block parasite growth with IC50 values from 1.9 to 7.0 µM, except for compound 32d (IC50 > 15 µM). Compared to compounds 32a–c, the extra lipophilic alkyl chain of compound 32d can limit its antimalarial activity.58) In 2015, Keough et al. reported some N-branched acyclic nucleoside phosphonates 33–35 as new PfHGXPRT inhibitors.59) These aza-acyclic nucleoside phosphonates contained uncommonly two phosphonate groups. The second phosphonate group was attached to the nitrogen atom through different linkers to fit into the PPi binding site. Compounds 33–35 were identified as potent inhibitors of PfHGXPRT with Ki values ranging from 0.19 to 10.0 µM (Fig. 6B), while their corresponding phosphoramidate prodrugs 36–38 were able to arrest parasite growth with IC50 values ranging from 1.0 to 6.0 µM. Inhibitors 36–38 were characterized by low cytotoxicity against human cells and may provide insights for the design of new antimalarial drug candidates.

The dissociation constants (Ki) for PfHGXPRT are colored red, and in vitro antimalarial activities against strains are colored blue.
Nucleoside analogues have a long and distinguished history in human therapy. The quest for new nucleoside analogues with high efficiency and low toxicity continues to be a fundamental approach to protect human health from fatal and debilitating diseases. Azanucleoside derivatives have gained much attention as sugar-modified nucleoside analogues because of their unique structures and versatile biological activities. Azanucleosides are reported to effectively treat cancer and show great promise in the treatment of other infections. The present mini-review summarizes the recent advances in the field of azanucleosides, where we focus on their molecular design and biological testing. The information provided on azanucleosides may be helpful to medicinal chemists in designing and exploiting novel chemical entities.
This work was funded by Doctor Scientific Research Startup Project Foundation of Xichang University, China (Grant No. YBZ202141), the Key Research and Development Program of Science and Technology Bureau of Liangshan Yi Autonomous Prefecture (Grant No. 21ZDYF0019). “Shuanggao” Science Research Program in Xichang University (Grant No. YBZ201904).
The authors declare no conflict of interest.