Experimental
General Experimental DetailsFor reactions that required heating, an oil bath was used as the heat source. Column chromatography was performed on Kanto Kagaku Silica Gel 60 N (spherical, neutral), 100–210 µm. When column chromatography required methanol as the eluent, silica gel was washed with methanol prior to use in order to remove inorganic salt impurity. Reactions and chromatography fractions were analyzed by TLC on a Wako Silica gel 70 F254 TLC Plate-Wako, with visualization by UV irradiation at 254 nm, phosphomolybdic acid, anisaldehyde, ninhydrin, and/or potassium permanganate staining. 1H-NMR spectra were recorded on a 400 MHz (100 MHz for 13C-NMR) JEOL JNM-ECZ-400S instrument. Chemical shifts and coupling constants are presented in ppm δ (relative to tetramethylsilane (0.00 ppm), CHCl3 (7.26 ppm), or CD2HOD (3.31 ppm)), and Hz, respectively. Chloroform-d1 (δ 77.16 ppm) and methanol-d4 (δ 49.00 ppm) were used as the internal standards for 13C-NMR spectroscopy. High-resolution MS (HR-MS) were obtained on a JEOL JMS-T100LP instrument for electrospray ionization (ESI). Fourier transform (FT)-IR spectra were recorded with a ThermoFisher Nicolet iS5 instrument with an iD5 attenuated total reflection (ATR) attachment and are reported in terms of frequency absorption (cm−1). All reagents were purchased from chemical companies and were used as received. Dehydrated solvents were purchased for the reactions and used without further desiccation, unless otherwise mentioned.
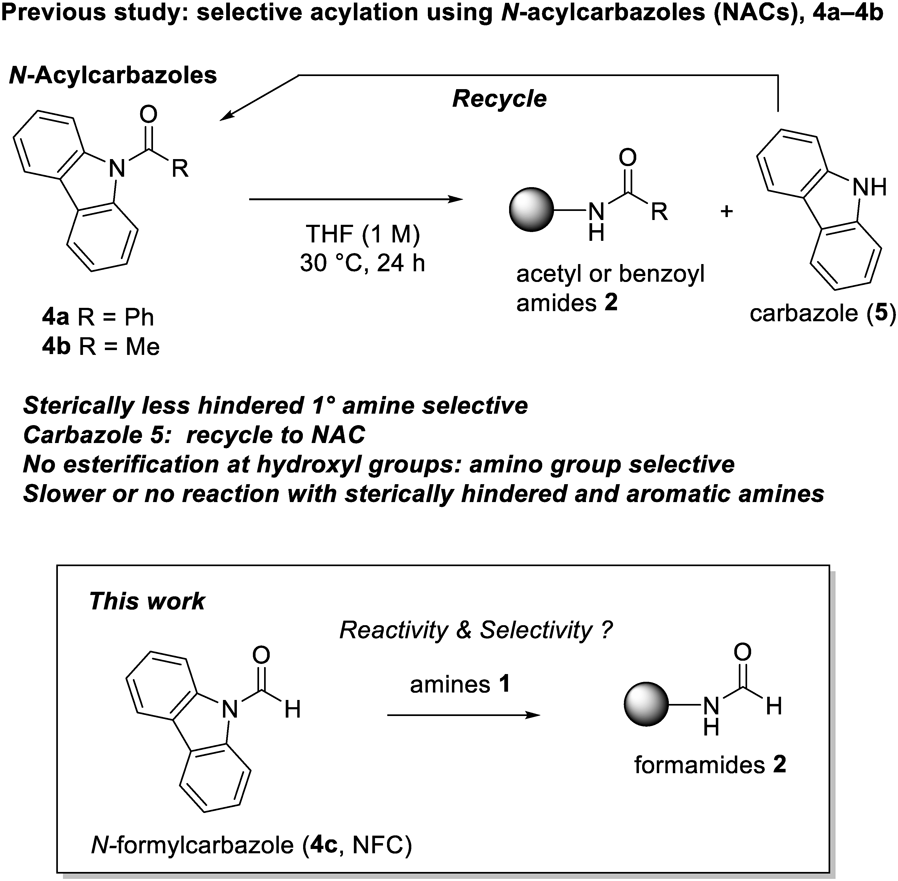
Preparation of N-Formylcarbazole (4c)N-Formylcarbazole 4c was prepared using a slightly modified version of the procedure described by Dixon and Lucas.20) A mixture of carbazole (50.0 g, 29.9 mmol) and formic acid (375 mL) was heated under reflux for 24 h. The reaction mixture was concentrated under reduced pressure. The resulting purple residue was dissolved in dichloromethane (200 mL). Activated charcoal (10.0 g) was then added to the mixture. The whole suspension was filtered through Celite, which was washed with dichloromethane (200 mL). The combined filtrate was concentrated under reduced pressure to give solids. Ether (200 mL) was then added, and the mixture was vigorously stirred for 1 h. The resulting suspension was filtered to give 4c as a white powder (43.8 g, 75%). The NMR data for 4c were identical with the reported values (see Supplementary materials for the spectrum).22)
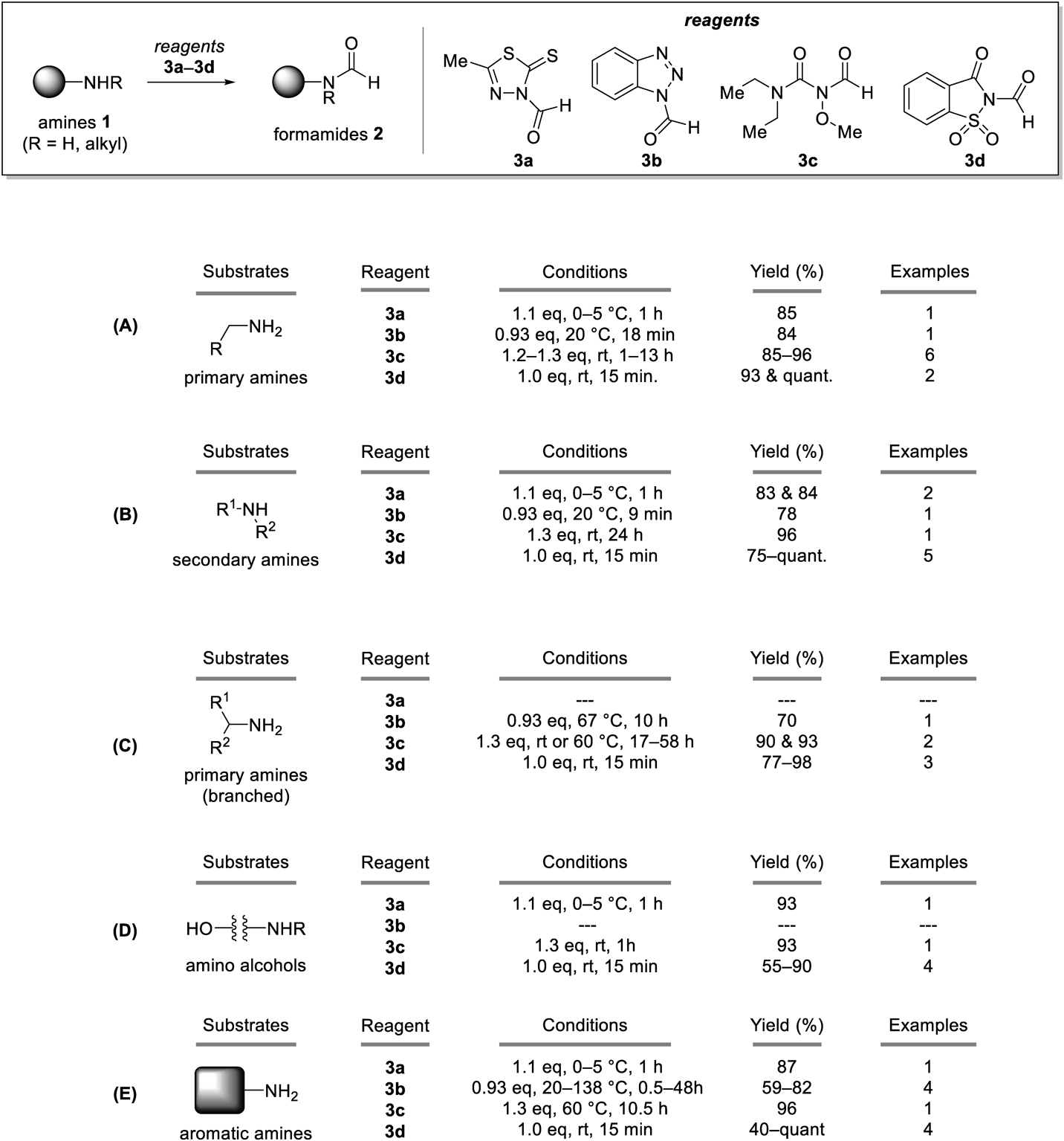
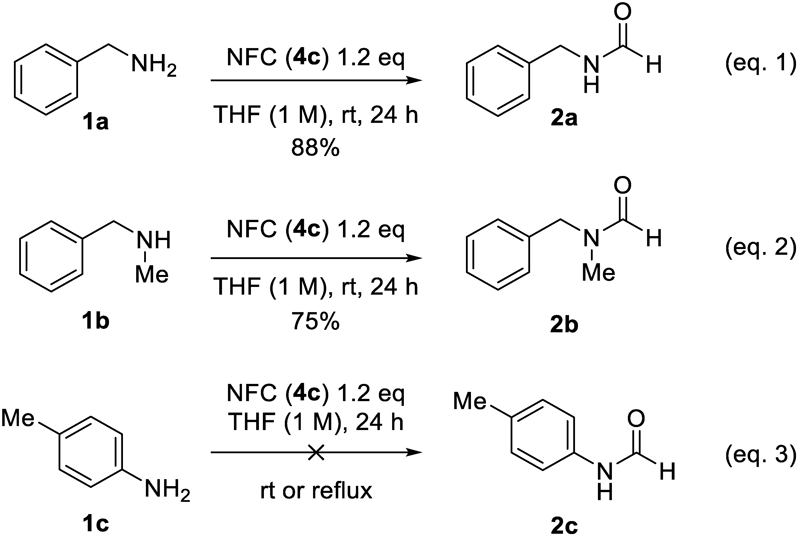
Typical Synthetic Procedure (Condition A)To a solution of N-formylcarbazole 4c (0.300 mmol) in THF (0.125 mL) was added amine (0.250 mmol) at 25 °C. After stirring for 24 h at 25 °C, the reaction mixture was diluted with dichloromethane (2.0 mL). The mixture was directly subjected to silica gel column chromatography. Carbazole, the stoichiometric byproduct was typically recovered in the first fractions eluted with dichloromethane.
Typical Synthetic Procedure (Condition B)To a solution of N-formylcarbazole 4c (0.600 mmol) in THF (0.125 mL) was added amine (0.250 mmol) at 25 °C. After refluxing for 24 h, the reaction mixture was cooled to room temperature and diluted with dichloromethane (2.0 mL). The mixture was directly subjected to silica gel column chromatography. Carbazole, the stoichiometric byproduct was typically recovered in the first fractions eluted with dichloromethane.
Typical Synthetic Procedure (Condition C)To a solution of N-formylcarbazole 4c (0.300 mmol) in THF (0.250 mL) was added amine (0.250 mmol) at 25 °C. After stirring for 12 h at 25 °C, the reaction mixture was diluted with dichloromethane (2.0 mL). The mixture was directly subjected to silica gel column chromatography. Carbazole, the stoichiometric byproduct was typically recovered in the first fractions eluted with dichloromethane.
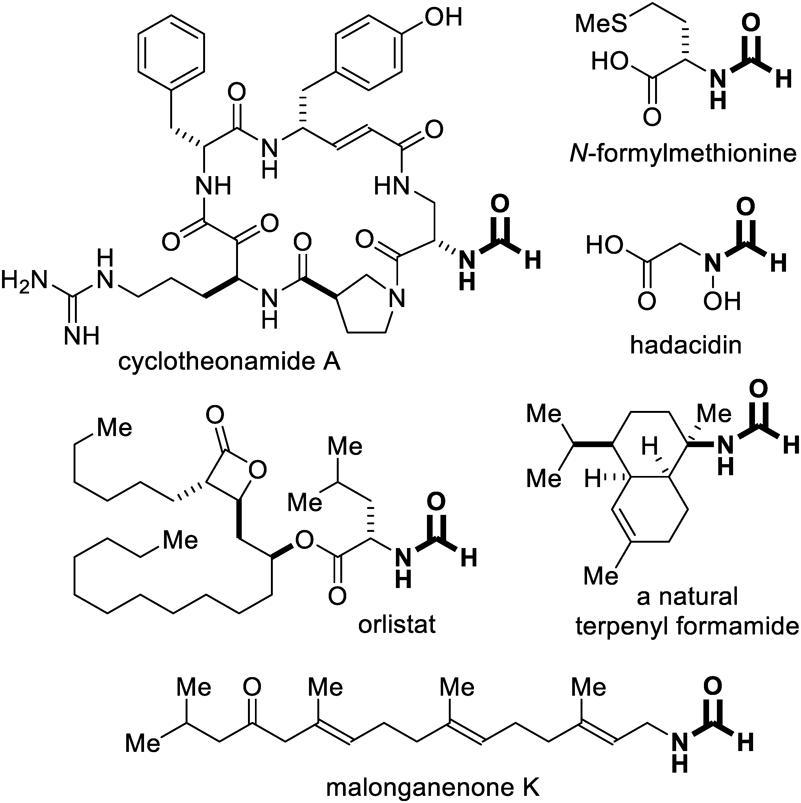
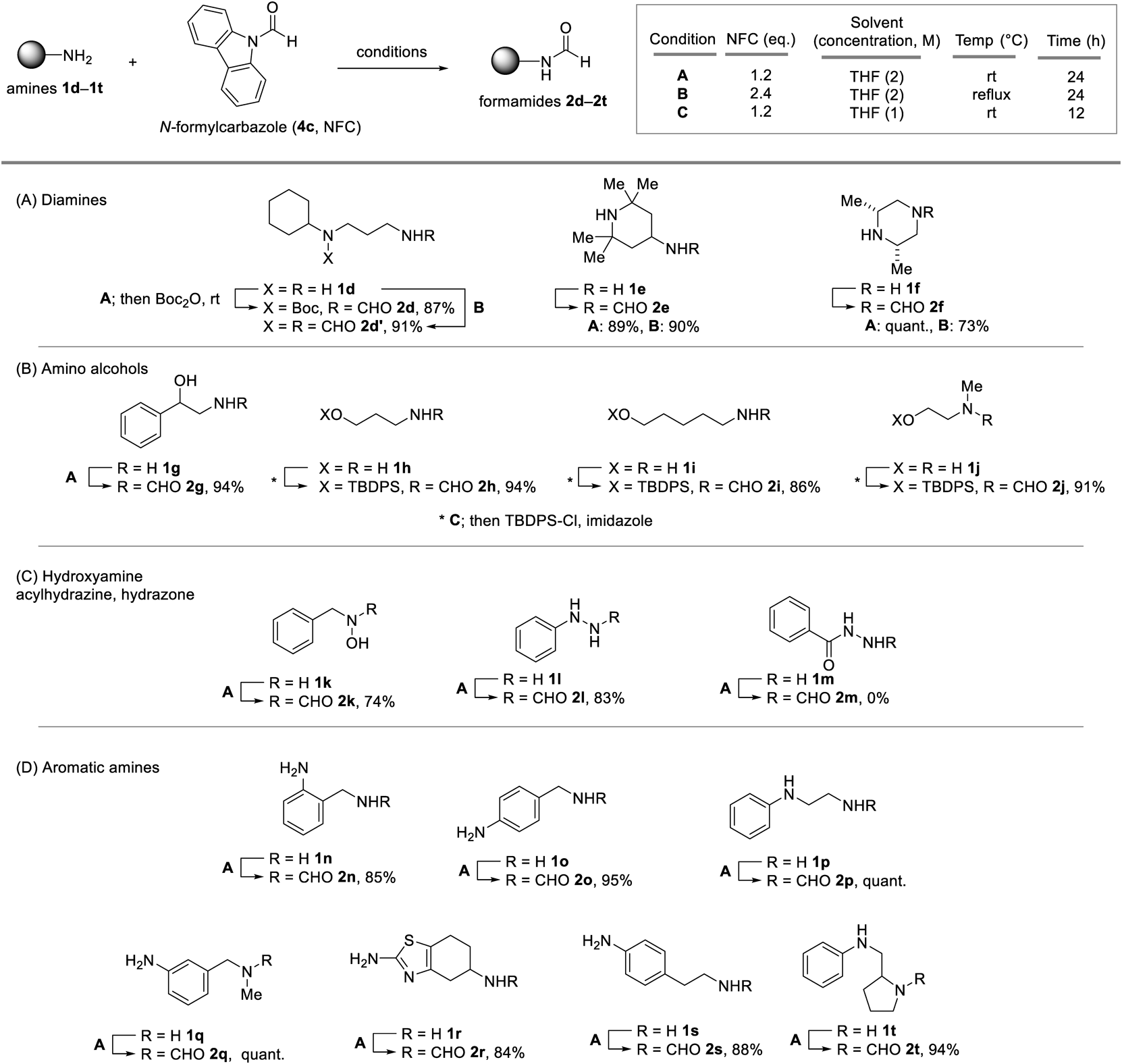
Synthesis and Characterization of CompoundsThe yields of 2a23) and 2b24) were determined by crude 1H-NMR spectroscopy using triphenylmethane as the internal standard. The NMR data for the synthetic formamides 2g,25)2k,26)2l,27)2n,28)2o,29) and 2p30) were identical to the literature values (see Supplementary materials for the spectra). The analytical data for the new compounds are as follows.
N1-tert-Butoxycarbonyl-N1-cyclohexyl-N3-formylpropane-1,3-diamine (2d)Following the formylation of 1d under “Condition A,” Boc2O (0.375 mmol) was added to the reaction mixture. After stirring for 1 h at 25 °C, the reaction mixture was diluted with dichloromethane (2.0 mL). The reaction mixture was directly subjected to silica gel column chromatography to obtain the title compound (62.0 mg, 87%) as colorless oil; 1H-NMR (CDCl3, 60 °C) δ: 8.16 (1H, s), 3.59 (1H, br s), 3.28–3.20 (4H, m), 1.79 (2H, d, J = 13.1 Hz), 1.71–1.62 (4H, m), 1.50–1.36 (9H + 3H, m), 1.33–1.24 (2H, m), 1.13–1.02 (1H, m); 13C-NMR (CDCl3) δ: 164.7, 161.5, 79.9, 57.1, 40.3, 34.3, 31.5, 30.0, 28.6, 26.2, 25.6; IR (ATR) cm−1: 3307, 3061, 2971, 2930, 2855, 1660, 1537, 1411, 1364, 1301, 1245; HR-MS m/z: 307.1998 (Calcd for C15H28N2Na2O3+: 307.1992).
N1-Cyclohexyl-N1-formyl-N3-formylpropane-1,3-diamine (2d′)The reaction under “Condition B” gave the title compound (48.3 mg, 91%) as pale yellow oil; 1H-NMR (CDCl3) δ: 8.21 (1H, s), 8.18 (1H, d, J = 1.2 Hz), 7.06 (1H, br s), 3.36 (2H, t, J = 6.5 Hz), 3.30–3.18 (3H, m), 1.92–1.77 (4H, m), 1.70 (3H, dt, J = 12.5, 6.5 Hz), 1.52 (2H, qd, J = 12.5, 3.5 Hz), 1.33 (2H, qt, J = 12.5, 3.5 Hz), 1.13 (1H, qt, J = 12.5, 3.5 Hz); 13C-NMR (CDCl3) δ: 163.5, 161.4, 58.9, 38.2, 34.2, 33.0, 29.9, 25.8, 25.1; IR (ATR) cm−1: 3292, 3052, 3052, 2931, 2855, 1651, 1532, 1468, 1449, 1426, 1383, 1355, 1296, 1235; HR-MS m/z: 235.1412 (Calcd for C11H20N2O2Na+: 235.1423).
N-(4-(2,2,6,6-Tetramethyl)piperidyl)formamide (2e)The reaction under “Condition A” with column chromatography (EtOAc then MeOH) gave the title compound (41.2 mg, 89%) as clear yellow oil; 1H-NMR (CDCl3) δ: 8.18 (0.2H, d, J = 12.0 Hz), 8.13 (0.8H, s), 5.39 (0.2NH, br s), 5.25 (0.8NH, br s), 4.41–4.31 (0.8H, m), 3.84–3.68 (0.2H, m), 1.92 (1.6H, dd, J = 12.8, 3.8 Hz), 1.85 (0.4H, dd, J = 12.8, 3.8 Hz), 1.26 (4.8H, s), 1.24 (1.2H, s), 1.15 (1.2H, s), 1.13 (4.8H, s), 1.04 (0.2H, t, J = 12.3 Hz), 0.95 (1.8H, t, J = 12.3 Hz); 13C-NMR (CDCl3, major rotamer) δ: 160.5, 51.1, 45.2, 41.5, 35.0, 28.6; 13C-NMR (CDCl3) δ: 163.7, 51.0, 46.9, 45.5, 34.9, 28.6; IR (ATR) cm−1: 3266, 3251, 3174, 2966, 2918, 2856, 1663, 1548, 1460, 1399, 1387, 1381, 1369, 1309, 1254, 1224; HR-MS m/z: 185.1654 (Calcd for C10H21N2O+ required 185.1648).
N4-Formyl-cis-2,6-dimethylpiperazine (2f)The reaction under “Condition A” gave the title compound (39.2 mg, 100%) as clear yellow oil; 1H-NMR (CDCl3) δ: 8.03 (1H, s), 4.29 (1H, d, J = 12.5 Hz), 3.42 (1H, d, J = 12.5 Hz), 2.93–2.75 (2H, m), 2.72 (1H, t, J = 11.7 Hz), 2.26 (1H, t, J = 11.7 Hz), 1.10 (6H, d, J = 6.1 Hz); 13C-NMR (CDCl3) δ: 160.8, 52.5, 51.9, 50.6, 46.68, 19.4, 19.3; IR (ATR) cm−1: 3493, 3294, 2965, 2905, 2860, 1658, 1439, 1397, 1317, 1259, 1216; HR-MS m/z: 143.1184 (Calcd for C7H15N2O+: 143.1179).
N-(3-tert-Butyldiphenylsiloxy)propylformamideAfter 1h was formylated under “Condition C” without column chromatography, silylation of the primary alcohol was conducted via a one-pot operation. Imidazole (0.200 mmol) and tert-butyldiphenylsilyl chloride (TBDPSCl) (0.375 mmol) were then added to the reaction mixture. After stirring for 48 h at 25 °C, the reaction was quenched with H2O. The mixture was extracted with EtOAc thrice, and the combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the title compound (80.6 mg, 94%) as colorless oil; 1H-NMR (CDCl3) δ: 8.06 (0.9H, s), 8.02 (0.1H, s), 7.71–7.61 (4H, m), 7.50–7.35 (6H, m), 5.98 (0.9NH, s), 5.74 (0.1NH, s), 3.78 (1.8H, t, J = 5.6 Hz), 3.73 (0.2H, t, J = 5.7 Hz), 3.45 (1.8H, q, J = 6.0 Hz), 3.40 (0.2H, q, J = 6.4 Hz), 1.81–1.67 (2H, m), 1.07 (9H, s); 13C-NMR (CDCl3, major rotamer) δ: 161.2, 135.7, 133.4, 130.2, 128.0, 62.8, 36.6, 31.4, 27.0, 19.3; 13C-NMR (CDCl3, minor rotamer) δ: 164.8, 135.7, 133.4, 130.2, 128.0, 60.9, 39.1, 33.5, 27.0, 19.3; IR (ATR) cm−1: 3287, 2930, 2857, 1669, 1111, 702; HR-MS m/z: 364.1696 (C20H27NO2SiNa+: 364.1709).
N-(5-tert-Butyldiphenylsilyloxy)pentylformamide (2i)After 1i was formylated under condition C,” the reaction mixture was diluted with dichloromethane (0.8 mL) and then, imidazole (0.200 mmol) and TBDPSCl (0.375 mmol) were added to the reaction mixture. After stirring for 24 h at 25 °C, the reaction was quenched with H2O. The mixture was extracted with EtOAc thrice, and the combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the title compound (63.9 mg, 86%) as colorless oil; 1H-NMR (CDCl3) δ: 8.14 (0.8H, s, 1H), 8.02 (0.2H, d, J = 12.0 Hz), 7.66 (4H, dd, J = 7.8, 1.5 Hz), 7.48–7.33 (6H, m), 5.45 (1NH, br s), 3.66 (2H, t, J = 6.3 Hz), 3.27 (1.6H, q, J = 6.6 Hz), 3.17 (0.4H, q, J = 6.7 Hz), 1.60–1.36 (6H, m), 1.05 (9H, s); 13C-NMR (CDCl3, major rotamer) δ: 161.2, 135.7, 134.2, 129.7, 127.77, 63.8, 38.3, 32.2, 29.3, 27.0, 23.3, 19.4; 13C-NMR (CDCl3, minor rotamer) δ: 164.6, 135.7, 134.1, 129.8, 127.78, 63.6, 41.8, 32.1, 31.1, 27.0, 22.9, 19.4; IR (ATR) cm−1: 2931, 2855, 1670, 1541, 1109, 702; HR-MS m/z: 392.2019 (Calcd for C22H31NO2SiNa+: 392.2022).
N-2-(tert-Butyldiphenylsiloxy)ethyl-N-methylformamide (2j)After 1j was formylated under “Condition C” without column chromatography, silylation of the primary alcohol was conducted via a one-pot operation. Imidazole (0.200 mmol) and TBDPSCl (0.375 mmol) were then added to the reaction mixture. After stirring for 48 h at 25 °C, the reaction was quenched with H2O. The mixture was extracted with EtOAc thrice, and the combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the title compound (78.0 mg, 91%) as colorless oil; 1H-NMR (CDCl3) δ: 8.08 (0.7H, s), 8.03 (0.3H, s), 7.69–7.59 (4H, m), 7.49–7.36 (6H, m), 3.81 (0.6H, t, J = 5.4 Hz), 3.70 (1.4H, t, J = 5.2 Hz), 3.48 (0.6H, t, J = 5.4 Hz), 3.33 (1.4H, t, J = 5.2 Hz), 3.02 (0.9H, s), 2.79 (2.1H, s), 1.05 (2.7H, s), 1.05 (6.3H, s); 13C-NMR (CDCl3, major rotamer) δ: 163.4, 135.62, 133.0, 130.0, 127.9, 60.7, 51.8, 30.1, 26.86, 19.15; 13C-NMR (CDCl3, minor rotamer) δ: 162.8, 135.61, 133.3, 129.9, 127.8, 62.0, 46.9, 36.3, 26.91, 19.20; IR (ATR) cm−1: 2930, 2857, 1672, 1428, 1111, 703; HR-MS m/z: 364.1692 (Calcd for C20H27NO2SiNa+: 364.1709).
N-[(3-Aminophenyl)methyl]-N-methylformamide (2q)The reaction under “Condition A” gave the title compound (41.0 mg, 100%) as clear pale brown oil; 1H-NMR (CDCl3) 8.23 (0.6H, s), 8.13 (0.4H, s), 7.14–7.07 (1H, m), 6.63–6.48 (3H, m), 4.41 (0.8H, s), 4.28 (1.2H, s), 3.78 (2NH, br s), 2.83 (1.2H, s), 2.77 (1.8H, s); 13C-NMR (CDCl3, major rotamer) δ: 162.9, 147.2, 137.0, 129.8, 117.3, 114.7, 113.5, 53.5, 29.5; 13C-NMR (CDCl3, minor rotamer) δ: 162.7, 147.0, 137.2, 129.5, 118.3, 114.6, 114.4, 47.6, 34.1; IR (ATR) cm−1: 3430, 3349, 3231, 3037, 3014, 2923, 2863, 1652, 1604, 1492, 1463, 1393, 1314, 1295, 1259, 1210; HR-MS m/z: 165.1002 (Calcd for C9H13N2O+: 165.1022).
N-Despropyl-N-formylprampexole (2r)The reaction under “Condition A” gave the title compound (41.6 mg, 84%) as clear pale brown oil; 1H-NMR (CD3OD) δ: 8.02 (1H, s), 4.60 (1NH, br s), 4.32–4.20 (1H, m), 2.88 (1H, dd, J = 15.5, 4.8 Hz), 2.58 (2H, dd, J = 7.5, 5.9 Hz), 2.48 (1H, ddt, J = 15.5, 7.5, 2.0 Hz), 2.06–1.93 (1H, m), 1.92–1.78 (1H, m); 13C-NMR (CD3OD) δ: 169.8, 163.3, 144.6, 114.5, 45.6, 29.8, 29.1, 24.9; IR (ATR) cm−1: 3296, 3192, 2925, 2851, 1662, 1522, 1370, 1307, 1272, 1237; HR-MS m/z: 198.0711 (C8H12N3OS+: 198.0696).
4-Aminophenethylformamide (2s)The reaction under “Condition A” gave the title compound (36.2 mg, 88%) as clear pale brown oil; 1H-NMR (CDCl3) δ: 8.06 (0.8H, s), 7.88 (0.2H, d, J = 12.0 Hz), 6.97 (1.6H, d, J = 8.3 Hz), 6.93 (0.4H, d, J = 8.3 Hz), 6.63 (2H, d, J = 8.3 Hz), 5.83 (1NH, br s), 3.63 (2NH, br s), 3.49 (1.6H, q, J = 6.7 Hz), 3.38 (0.4H, q, J = 6.7 Hz), 2.76–2.62 (2H, m); 13C-NMR (CDCl3, major rotamer) δ: 164.6, 145.3, 129.6, 128.3, 115.46, 39.5, 34.6; 13C-NMR (CDCl3, minor rotamer) δ: 161.3, 145.1, 129.7, 128.3, 115.54, 43.5, 36.9; IR (ATR) cm−1: 3337, 3234, 3050, 3033, 3018, 3004, 2934, 2860, 1660, 1516, 1453, 1385, 1275, 1237; HR-MS m/z: 187.0874 (Calcd for C9H12N2NaO+: 187.0842).
N1-Formyl-2-anilinomethylpyrrolidine (2t)The reaction under “Condition A” gave the title compound (47.9 mg, 94%) as clear pale brown oil; 1H-NMR (CDCl3) δ: 8.26 (0.5H, s), 8.21 (0.5H, s), 7.19–7.13 (2H, m), 6.75–6.57 (3H, m), 4.37–4.31 (0.5H, m), 4.08–4.02 (0.5H, m), 3.65–3.53 (1H, m), 3.48–3.35 (1H, m), 3.30–3.04 (2H, m), 2.18–1.72 (4H, m); 13C-NMR (CDCl3, major rotamer) δ: 162.5, 148.3, 129.2, 117.0, 112.5, 55.1, 48.6, 476.9, 29.4, 23.9; 13C-NMR (CDCl3, minor rotamer) δ: 161.5, 147.1, 129.5, 118.0, 112.8, 56.1, 48.1, 43.5, 28.5, 22.5; IR (ATR) cm−1: 3348, 3051, 3026, 2970, 2877, 1651, 1601, 1506, 1499, 1417, 1382, 1319, 1259; HR-MS m/z: 205.1343 (Calcd for C12H17N2O+: 205.1335).