Regular Articles
p-Methoxy- and 2,4-Dimethoxybenzyl N-Acetylcarbamate Potassium Salts: Versatile Reagents for N-Alkylacetamide and p-Methoxy- and 2,4-Dimethoxybenzyl Carbamates
2022 年 70 巻 9 号 p. 650-661
詳細
2022 年 70 巻 9 号 p. 650-661
The acetamide moiety is a general functional group present in many natural and pharmaceutical products. Herein, we report two new reagents, p-methoxybenzyl N-acetylcarbamate potassium salt (PM-BENAC-K) and 2,4-dimethoxybenzyl N-acetylcarbamate potassium salt (2,4-DM-BENAC-K), which are refined versions of the benzyl N-acetylcarbamate potassium salt (BENAC-K). These compounds, which we reported as simple equivalents of N-acetamide nucleophiles, are stable and easy-to-handle powders that react with a variety of alkyl halides and sulfonates to afford substituted products in good yields. The products were transformed into N-alkylacetamides after p-methoxybenzyloxycarbonyl (Moz) or 2,4-dimethoxybenzyloxycarbonyl (Dmoz) cleavage under mild acidic conditions. The acetyl groups in the substituted products of PM- and 2,4-DM-BENAC-Ks were removed using K2CO3 in methanol to afford Moz- and Dmoz-protected amines, respectively. Hence, the new BENAC-Ks acted as versatile equivalents of both N-acetamide and Moz/Dmoz-protected nitrogen nucleophiles and can be used in synthetic studies of natural and pharmaceutical products.
Acetamide is a representative carboxamide functional group used in organic chemistry. In nature, acetamide moieties are present in various physiological compounds and complex natural products (Fig. 1A). For example, melatonin is a natural hormone that controls biological circadian rhythms in living organisms. Aspongamide A, produced by the Chinese pentatomidae insect, Aspongopus chinensis, includes three acetamide moieties per molecule.1) Brevisamide2,3) and tamulamides4) are notable polyketides with an acetamide moiety, which have been isolated from the red-tide dinoflagellate, Karenia brevis. Acetamide moieties are also present in many pharmaceutical products, such as the antidepressant agomelatine, which acts as an agonist for melatonin MT1 and MT2 receptors and an antagonist for serotonin 5-HT2c receptors,5,6) and antibiotic linezolide developed for multidrug-resistant Gram-positive bacteria.7,8) When the drug linezolid was discovered, the acetamide functional group was found to play an important role in enhancing biological activities. Alkyl halides are common precursors of N-alkylacetamides, and direct nucleophilic substitution with acetamide is the most straightforward method for the synthesis of N-alkylacetamides from alkyl halides (Fig. 1B). However, this is typically difficult because the solubility of acetamide in organic solvents is low, and a large excess of acetamide is necessary to avoid the formation of dialkylated byproducts.9–15) Therefore, a three-step transformation involving azidation, reduction, and acetylation is often employed,16) although this procedure is disadvantageous owing to the formation of an explosive alkyl azide intermediate.

To address the aforementioned issue, we recently developed a benzyl N-acetylcarbamate potassium salt (BENAC-K) as a simple and useful reagent for the construction of N-alkylacetamide moieties from alkyl halides and alkyl sulfonates17–19) (Fig. 2A). An SN2 reaction20–28) of an alkyl halide with BENAC-K provided substituted products, in which the benzyloxycarbonyl (Cbz) group was removed by hydrogenation to afford N-alkylacetamide products. In contrast, when the substituted product was treated under basic conditions, the acetyl group was preferentially cleaved to afford Cbz-protected amines. Next, we designed advanced variants of BENAC-K: p-methoxybenzyl N-acetylcarbamate potassium salt (PM-BENAC-K, 1a) and 2,4-dimethoxybenzyl N-acetylcarbamate potassium salt (2,4-DM-BENAC-K, 1b). Because the p-methoxybenzyloxycarbonyl (Moz) group is slightly more sensitive to acidic conditions than the tert-butoxycarbonyl (Boc) group,29,30) we assumed that the alkylated products would be transformed into N-alkyl acetamide under mild acidic conditions. Furthermore, the substituted products are expected to be converted to Moz- and 2,4-dimethoxybenzyloxycarbonyl (Dmoz)-protected amines after base-mediated deacetylation. The Moz group is a useful protecting group for amino acids in peptide synthesis,31,32) and Moz protection is also advantageous for the protection of amines that do not exhibit strong absorption at 254 nm (Fig. 2B). However, Moz is used less frequently than Cbz and Boc because reagents for Moz protection, such as p-methoxybenzyl azide,33,34) p-methoxybenzyl S-(4,6-dimethylpyrimidin-2-yl)thiocarbonate,35) and 2-(4-methoxybenzyloxycarbonyloxyimino)-2-phenylacetonitrile36) are expensive compared to the protection reagents for Cbz or Boc. Notably, Dmoz-protected amines are unexplored because reagents for protection with Dmoz have not yet been prepared. Therefore, we speculated that the alkylated products of PM- and 2,4-DM-BENAC-Ks would be useful as versatile reagents for the synthesis of Moz- and Dmoz-protected amines as well as for the introduction of N-acetamide. Herein, we report the synthesis and alkylation reaction of PM-BENAC-K and 2,4-DM-BENAC-K and the subsequent selective deprotection of N-alkylacetamides and Moz/Dmoz-protected amines.

The syntheses of PM-BENAC-K and 2,4-DM-BENAC-K were performed using a three-step procedure involving methyl carbamate (2, Chart 1). N-Acetylation using acetic anhydride and Amberlyst 15 afforded methyl N-acetylcarbamate (3). The initial attempt at thermal transesterification with p-methoxybenzyl alcohol in xylene37,38) through acetyl isocyanate failed owing to the formation of di(p-methoxybenzyl) ether.39) Significantly, the addition of a stoichiometric quantity of i-Pr2NEt suppressed the formation of the ethereal side product, and the desired p-methoxybenzyl N-acetylcarbamate (4a) was obtained in an excellent yield.40) This condition can also be applied to transesterification using 2,4-dimethoxybenzyl alcohol to afford 2,4-dimethoxybenzyl N-acetylcarbamate (4b). The subsequent treatment of 4a and 4b with potassium tert-butoxide (t-BuOK) afforded PM- and 2,4-DM-BENAC-K (1a–b), respectively, as white powders. Notably, the purification of all three conversions was performed only by recrystallization, and the preparation of both PM- and 2,4-DM-BENAC-K was easily scaled up to over 10 g in our laboratory. Both BENAC-Ks were stable in a freezer (−25 °C) for more than two years.41) Their stability is sufficient for practical use, in contrast to that of t-butyl acetylcarbamate potassium salt (Boc(Ac)N−K+), which could not be developed as a reagent because of its hygroscopic nature.18)

Reactions using PM- and 2,4-DM-BENAC-Ks (1a–b) were performed in dimethylformamide (DMF) without additives according to the optimized reaction conditions in our previous reports18) for the original BENAC-K (Fig. 3), and the substrate scope for the substitution reactions using 1a–b was comparable to that of BENAC-K. For example, a variety of primary alkyl halides, including benzyl 3-bromopropyl ether, lauryl bromide, isobutyl bromide, and cyclohexyl bromide, were subjected to alkylation in DMF at 60–80 °C (entries 1–8). Reactive alkyl halides, such as benzyl bromide, allyl bromide, propargyl bromide, and ethyl bromoacetate, underwent SN2 substitution smoothly at room temperature (r.t.) or 0 °C (entries 9–16). Alkyl chloride, iodide, and tosylate functioned as good alkyl sources for PM- and 2,4-DM-BENAC-K (entries 19–24). Because the substitution reaction proceeds via an SN2 mechanism, secondary alkyl halides are more challenging alkylating reagents than primary alkyl halides. Experiments with 2-bromopropionate under standard conditions in DMF resulted in a poor yield because of the formation of O-alkylated side products. The formation of the O-alkylated byproduct was also challenging when alkyl triflate,42) the most cationic electrophile, was used (Fig. 3). Significantly, the formation of O-alkylated byproducts was suppressed to provide alkylated products 13a–b and 15a–b in practical yields when reactions were conducted in a less-polar solvent using 18-crown-6 (entries 17, 18, 25, and 26).18) We speculated that the reactivity of a “hard” oxygen nucleophile diminished because the alkyl halide and triflate were less polarized in apolar solvents.

The demasking of Cbz, Moz, and Dmoz under acidic conditions was demonstrated (Fig. 4a). The Cbz group of 16, which is a product of 3-bromopropyl benzyl ether and BENAC-K,18) was removed by trifluoroacetic acid at room temperature. However, 16 was unreactive under milder acidic conditions using formic acid, which readily cleaved the Moz group of 5a after 1 h. The Dmoz group was sensitive in less acidic conditions, such as acetic acid in CH2Cl2 or a catalytic quantity of TsOH·H2O in MeOH-CH2Cl2 co-solvent. Therefore, the order of sensitivity to Brønsted acids was Cbz < Moz < Dmoz. Next, we examined the other general conditions used for the deprotection of p-methoxybenzyl ethers (Fig. 4b). The removal of the Dmoz group in 5b was also achieved through oxidation using ceric ammonium nitrate (CAN) in CH3CN, while the product yield was moderate. In other cases, oxidative conditions were ineffective for deprotection. The Moz moiety in 5a was retained under CAN oxidation conditions, and 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) did not achieve deprotection of either 5a or 5b at room temperature. Hydrogenolysis catalyzed by Pd/C was used successfully for the removal of the Moz group in 5a, and Moz was removed in advance of the benzyl ether. The high reactivity of Moz toward hydrogenation is similar to that of the Cbz group in a previous report.18) The selective removal of acetyl groups was demonstrated using α-amino acid derivatives 12a–b and 13a–b (Fig. 4c). The methanolysis of 12a–b with K2CO3 in methanol prompted both deacetylation and transesterification to afford Moz- and Dmoz-protected glycine methyl esters 18a–b in good yields. Finally, the deacetylation of 13a–b provided Moz- and Dmoz-protected alanine methyl esters 19a–b.

We developed two new reagents, PM-BENAC-K and 2,4-DM-BENAC-K, which are versatile sources of N-acetamide anions and p-methoxy- and 2,4-dimethoxybenzyl carbamate N-anions. Both reagents are stable powders that can be easily handled in an air atmosphere. A variety of alkyl halides and sulfonates underwent substitution reactions with PM- and 2,4-DM-BENAC-Ks, providing substituted products in good yields. The treatment of the products under mildly acidic conditions cleaved Moz, and the Dmoz group provided N-alkylacetamides. In addition, Moz- and Dmoz-protected amines were obtained through treatment with K2CO3 in methanol. PM- and 2,4-DM-BENAC-K are expected to be useful sources of N-alkylacetamides and Moz/Dmoz-protected amines for synthetic studies of natural and pharmaceutical products.
All air- and moisture-sensitive reactions were carried out under an argon atmosphere in commercially available dry solvents under anhydrous conditions. Heating reactions were performed using an oil bath otherwise noted. Flash chromatography was carried out using silica gel (spherical, neutral, particle size 40–50 µm, Kanto Chemical, Tokyo, Japan). Specific optical rotations were recorded on a JASCO P-1020 polarimeter. IR spectra were recorded on JASCO FT/IR-410 or JASCO FT/IR-4600 spectrophotometers. NMR spectra were recorded on AVANCE III HD (600 MHz), JNM-ECA (500 MHz), or JNM-ECZS (400 MHz) spectrometers. Chemical shifts are reported in ppm relative to the solvent signals (δ 2.50 ppm for dimethyl sulfoxide (DMSO)-d6) or internal TMS (δ 0.00 ppm for CDCl3) for 1H-NMR spectra and to the solvent signals (δ 39.51 ppm for DMSO-d6, δ 77.0 ppm for CDCl3) for 13C-NMR spectra. Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad). High-resolution mass spectra were recorded on an Exactive Plus orbitrap mass spectrometer (direct analysis in real time (DART), electrospray ionization (ESI)).

A mixture of methyl carbamate (2) (10.0 g, 133.2 mmol, 1.0 equivalent (equiv.)) and amberlyst 15 (1.0 g) in acetic anhydride (25.0 mL, 27.0 g, 262 mmol, 2.0 equiv.) was stirred at room temperature for 3 h. The mixture was filtered with aid of EtOAc and the filtrate was concentrated under reduced pressure. The residual solid was purified by recrystallization from toluene (20 mL)-n-hexane (70 mL) to afford methyl N-acetylcarbamate (3) (14.0 g). The concentrate of mother liquid was subjected to the second recrystallization to afford additional 3 (0.79 g). In total, 14.8 g (95%) of 3 was obtained as a colorless needles of mp 95–96 °C; IR (KBr pellet) 3266, 3192, 1771, 1508, 1211 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 8.31 (1H, br s), 3.79 (3H, s), 2.45 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.9, 152.6, 53.0, 23.9; Melting point was identical to literature data (93 °C).43)

A 500-mL round-bottomed flask equipped with a magnetic stir bar and a distillation apparatus was charged with a solution of methyl N-acetylcarbamate (3) (10.0 g, 85.4 mmol), p-methoxybenzyl alcohol (12.6 mL, 101 mmol, 1.18 equiv.), i-Pr2NEt (18.0 mL, 105 mmol, 1.23 equiv.), and xylenes (200 mL). The mixture was stirred under refluxing temperature (heating with aluminum block heater at 170 °C) in the manner of distillation for removal of methanol. After 3 h, additional xylenes (200 mL) were added and the distillation was continued for additional 3 h. After cooling to room temperature, n-hexane (150 mL) was added and the precipitate was collected with suction filtration. Recrystallization from toluene (50 mL)–n-hexane (150 mL) afforded 4a (17.0 g, 89%) as colorless needles of mp 119–120 °C; IR (film) 3204, 3143, 2983, 1692, 1679, 1503, 1234 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.69 (1H, br s), 7.30 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.6 Hz, JAX′ = 0 Hz), 6.90 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.6 Hz, JAX′ = 0 Hz), 5.11 (2H, s), 3.81 (3H, s), 2.42 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.1, 159.9, 151.8, 130.3, 127.0, 114.0, 67.7, 55.3, 24.0; HRMS (DART/Orbitrap) m/z: [M + NH4]+ Calcd for C11H17N2O4+ 241.1183; Found 241.1183.

A 500-mL round-bottomed flask equipped with a magnetic stir bar and a distillation apparatus was charged with a solution of methyl N-acetylcarbamate (3) (10.6 g, 90.7 mmol), 2,4-dimethoxybenzyl alcohol (18.3 g, 109 mmol, 1.2 equiv.), i-Pr2NEt (19.0 mL, 109 mmol, 1.2 equiv.), and xylenes (200 mL). The mixture was stirred under refluxing temperature (heating with aluminum block heater at 181 °C) for 3 h in the manner of distillation for removal of methanol. After cooling to room temperature, n-hexane (150 mL) was added and the precipitate was collected with suction filtration. Recrystallization from toluene (50 mL)–n-hexane (150 mL) afforded 4b (18.2 g, 80%) as colorless needles of mp 127–129 °C; IR (KBr pellet) 3215, 3154, 2996, 1752, 1696, 1510, 1220 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.45 (1H, br s), 7.25 (1H, ABX, JAB = 2.1 Hz, JAX = 8.4 Hz, JBX = 0 Hz, νAB = 0.4 Hz), 6.47 (2H, ABX, JAB = 2.1 Hz, JAX = 8.4 Hz, JBX = 0 Hz, νAB = 0.4 Hz), 5.16 (2H, s), 3.83 (3H, s), 3.82 (3H, s), 2.42 (3H, s); 13C{1H} NMR (125 MHz, CDCl3) δ: 171.9, 161.7, 159.1, 151.9, 131.9, 115.6, 104.1, 98.6, 63.5, 55.5, 55.4, 23.9; HRMS (DART/Orbitrap) m/z: [M–H]– Calcd for C12H14NO5− 252.0877; Found 252.0878.

To a solution of phenyl carbamate (20.0 g, 146 mmol) in acetic anhydride (200 mL, 2.12 mol, 14.5 equiv.) was added amberlyst 15 (2.0 g), and the reaction mixture was stirred at room temperature for 2 h. The mixture was filtered with aid of toluene, and the filtrate was concentrated under reduced pressure. The residue was purified by recrystallization from toluene (250 mL)-hexane (180 mL) to afford phenyl N-acetylcarbamate (23.1 g, 88%) as colorless needles of mp 127.6–128.3 °C; IR (KBr pellet) 3255, 3195, 3006, 1768, 1530, 1174 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 8.30 (1H, br s), 7.42–7.38 (2H, m), 7.27 (1H, m), 7.16–7.14 (2H, m), 2.49 (3H, s); 13C-NMR (125 MHz, CDCl3) δ: 172.6, 150.7, 149.8, 129.5, 126.4, 121.4, 24.1; Melting point was identical to literature data (123 °C).43)
Preparation of 4a from Phenyl N-AcetylcarbamateA solution of phenyl N-acetylcarbamate (10.0 g, 55.8 mmol), p-methoxybenzyl alcohol (8.31 mL, 67.0 mmol, 1.2 equiv.) and Et3N (9.31 mL, 67.0 mmol, 1.2 equiv.) in toluene (56 mL) was stirred at 80 °C for 1 h. After cooling to room temperature, n-hexane (150 mL) was added and the precipitates were collected with suction filtration. Recrystallization from toluene (100 mL)–n-hexane (100 mL) afforded 4a (9.57 g, 77%).
Preparation of 4b from Phenyl N-AcetylcarbamateA solution of phenyl N-acetylcarbamate (3.20 g, 17.8 mmol), 2,4-dimethoxybenzyl alcohol (3.60 g, 21.4 mmol), and Et3N (2.84 mL, 21.4 mmol) in toluene (19 mL) was stirred at 80 °C for 1 h. After cooling to room temperature, n-hexane (50 mL) was added and the precipitates were collected with suction filtration. Recristalization from toluene (35 mL)-n-hexane (30 mL) afforded 4b (3.60 g, 80%).

To a solution of 4-methoxybenzyl N-acetylcarbamate (4a) (18.6 g, 83.5 mmol, 1.0 equiv.) in 1,2-dimethoxyethane (150 mL) was added t-BuOK (9.4 g, 83.5 mmol, 1.0 equiv.), and the reaction mixture was stirred at room temperature for 10 min. The resulting precipitates were collected by suction filtration and washed with Et2O. The obtained white powder was dried under reduced pressure (25 °C, 2 mmHg) overnight to afford PM-BENAC-K (1a) (21.2 g, 97%) as colorless powder. Alkylation reactions were performed using the powder of PM-BENAC-K without further purification. mp 245–247 °C; IR (KBr pellet) 2996, 1679, 1567, 1225 cm−1; 1H-NMR (600 MHz, DMSO-d6) δ: 7.24 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.6 Hz, JAX′ = 0 Hz), 6.87 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.6 Hz, JAX′ = 0 Hz), 4.78 (2H, s), 3.73 (3H, s), 1.81 (3H, s); 13C{1H} NMR (150 MHz, DMSO-d6) δ: 178.3, 161.4, 158.4, 130.9, 129.0, 113.5, 64.1, 55.0, 26.6; HRMS (ESI/Orbitrap) m/z: [M–K]– Calcd for C11H12NO4– 222.0772; Found 222.0773.

To a solution of 2,4-dimethoxybenzyl N-acetylcarbamate (4b) (18.1 g, 71.9 mmol, 1.0 equiv.) in 1,2-dimethoxyethane (160 mL) was added t-BuOK (8.02 g, 71.9 mmol, 1.0 equiv.), and the reaction mixture was stirred at room temperature for 10 min. The resulting precipitates were collected by suction filtration and washed with Et2O. The obtained white powder was dried under reduced pressure (25 °C, 2 mmHg) overnight to afford 2,4-DM-BENAC-K (1b) (20.0 g, 99%) as colorless powder. Alkylation reactions were performed using the powder of 2,4-DM-BENAC-K without further purification. mp 184–187 °C; IR (KBr pellet) 2998, 2966, 2934, 2878, 2836, 1684, 1562, 1222 cm−1; 1H-NMR (500 MHz, DMSO-d6) δ: 7.18 (1H, d, J = 8.5 Hz), 6.52 (1H, d, J = 2.3 Hz), 6.47 (1H, dd, J = 8.5, 2.3 Hz), 4.76 (2H, s), 3.76 (3H, s), 3.74 (3H, s), 1.80 (3H, s); 13C{1H} NMR (125 MHz, DMSO-d6) δ: 178.4, 161.7, 159.9, 157.7, 129.4, 118.9, 104.2, 98.0, 59.6, 55.3, 55.2, 26.7; HRMS (ESI/Orbitrap) m/z: [M–K]– Calcd for C12H14NO5– 252.0877; Found 252.0878.

To a solution of benzyl 3-bromopropyl ether (232 mg, 1.01 mmol) in DMF (1 mL) was added PM-BENAC-K (1a) (287 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 3 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (15→30% EtOAc in n-hexane) afforded 5a (384 mg, quantative yield) as a colorless oil. IR (film) 2957, 2859, 1735, 1699, 1197 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.34–7.27 (7H, m), 7.24 (2H, AA′XX′, JAA′ = 2.6 Hz, JXX′ = 2.6 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.15 (2H, s), 4.43 (2H, s), 3.85 (2H, AA′XX′, JAA′ = 10.0 Hz, JXX′ = 10.0 Hz, JAX = 8.5 Hz, JAX′ = 6.0 Hz), 3.79 (3H, s), 3.47 (2H, t, J = 6.2 Hz), 2.47 (3H, s), 1.83 (2H, m); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.9, 159.8, 154.6, 138.4, 130.1, 128.3, 127.53, 127.46, 127.2, 114.0, 72.7, 68.3, 68.1, 55.2, 41.8, 28.8, 26.8; HRMS (DART/Orbitrap) m/z: [M + NH4]+ Calcd for C21H29N2O5+ 389.2071; Found 389.2067.

To a solution of benzyl 3-bromopropyl ether (111 mg, 0.48 mmol) in DMF (3 mL) was added 2,4-DM-BENAC-K (1b) (154 mg, 0.53 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (50% Et2O in n-hexane) afforded 5b (152 mg, 79%) as a colorless oil. IR (film) 2958, 1734, 1196 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.33–7.24 (6H, m), 6.44 (1H, d, J = 2.3 Hz), 6.42 (1H, dd, J = 8.5, 2.3 Hz), 5.19 (2H, s), 4.41 (2H, s), 3.83 (2H, AA′XX′, JAA′ = 10.0 Hz, JXX′ = 10.0 Hz, JAX = 8.5 Hz, JAX′ = 6.0 Hz), 3.79 (3H, s), 3.78 (3H, s), 3.46 (2H, t, J = 6.2 Hz), 2.46 (3H, s), 1.83 (2H, m); 13C{1H} NMR (125 MHz, CDCl3) δ: 173.0, 161.6, 159.1, 154.7, 138.5, 131.6, 128.3, 127.5, 127.4, 115.8, 103.9, 98.5, 72.6, 68.1, 64.2, 55.34, 55.29, 41.7, 28.6, 26.7; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C22H27NNaO6+ 424.1731; Found 424.1734.

To a solution of lauryl bromide (249 mg, 1.00 mmol) in DMF (5.5 mL) was added PM-BENAC-K (1a) (287 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 1.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (15% EtOAc in n-hexane) afforded 6a (369 mg, 94%) as a colorless oil. IR (film) 2925, 2854, 1736, 1703 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.32 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 6.90 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.16 (2H, s), 3.82 (3H, s), 3.68 (2H, t, J = 7.7 Hz), 2.48 (3H, s), 1.47 (2H, m), 1.31–1.22 (18H, m), 0.88 (3H, t, J = 7.0 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.8, 159.9, 154.7, 130.1, 127.2, 114.0, 68.2, 55.2, 44.2, 31.9, 29.63, 29.61, 29.56, 29.5, 29.3, 29.2, 28.6, 26.9, 26.8, 22.7, 14.1; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C23H37NNaO4+ 414.2615; Found 414.2618.

To a solution of lauryl bromide (125 mg, 0.50 mmol) in DMF (3 mL) was added 2,4-DM-BENAC-K (1b) (163 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (10% EtOAc in n-hexane) afforded 6b (172 mg, 81%) as a colorless oil. Colorless solid; mp 48–50 °C; IR (film) 2925, 2854, 1733, 1699, 1210, 1160 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.25 (1H, m), 6.48–6.45 (2H, m), 5.19 (2H, s), 3.82 (6H, s), 3.67 (2H, t, J = 7.8 Hz), 2.47 (3H, s), 1.47 (2H, m), 1.32–1.21 (18H, m), 0.88 (3H, t, J = 7.0 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 173.0, 161.6, 159.1, 154.9, 131.6, 115.9, 103.9, 98.5, 64.1, 55.4, 55.3, 44.2, 31.9, 29.64, 29.63, 29.59, 29.5, 29.34, 29.29, 28.5, 26.8 (x2), 22.7, 14.1; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C24H39NNaO5+ 444.2720; Found 444.2727.

To a solution of isobutyl bromide (110 µL, 1.00 mmol) in DMF (1 mL) was added PM-BENAC-K (1a) (288 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at 80 °C for 23.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 7a (167 mg, 60%) as a colorless oil. IR (film) 2961, 1734, 1702, 1227, 1150 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.32 (2H, AA′XX′, JAA′ = 2.6 Hz, JXX′ = 2.6 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 6.91 (2H, AA′XX′, JAA′ = 2.6 Hz, JXX′ = 2.6 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.15 (2H, s), 3.82 (3H, s), 3.57 (2H, d, J = 7.4 Hz), 2.49 (3H, s), 1.91 (1H, t-septet, J = 7.4, 6.8 Hz), 0.82 (6H, d, J = 6.8 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 173.0, 159.8, 154.9, 130.2, 127.1, 113.9, 68.2, 55.2, 50.6, 27.7, 26.8, 19.9; HRMS (DART/Orbitrap) m/z: [M + NH4]+ Calcd for C15H25N2O4+ 297.1809; Found 297.1812.

To a solution of isobutyl bromide (54.4 µL, 0.50 mmol) in DMF (1 mL) was added 2,4-DM-BENAC-K (1b) (162 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 18.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 7b (132 mg, 85%) as a colorless oil. IR (film) 2960, 1734, 1226, 1154 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 7.24 (1H, m), 6.48–6.45 (2H, m), 5.18 (2H, s), 3.820 (3H, s), 3.817 (3H, s), 3.55 (1H, d, J = 7.3 Hz), 2.48 (3H, s), 1.91 (1H, t-septet, J = 7.3, 6.9 Hz), 0.82 (6H, d, J = 6.9 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 173.2, 161.6, 159.1, 155.2, 131.7, 115.8, 103.8, 98.4, 64.2, 55.3, 55.2, 50.7, 27.6, 26.7, 19.9; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C16H23NNaO5+ 332.1468; Found 332.1470.

To a solution of (bromomethyl)cyclohexane (140 µL, 1.00 mmol) in DMF (1 mL) was added PM-BENAC-K (1a) (288 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 19.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 8a (259 mg, 81%) as a colorless oil. IR (film) 2925, 2851, 1733, 1701, 1515, 1203 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.32 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 6.91 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.16 (2H, s), 3.82 (3H, s), 3.58 (2H, d, J = 6.8 Hz), 2.48 (3H, s), 1.69–1.51 (6H, m), 1.15–1.08 (3H, m), 0.93–0.85 (2H, m); 13C{1H} NMR (150 MHz, CDCl3) δ: 173.0, 159.8, 155.0, 130.2, 127.2, 113.9, 68.2, 55.2, 49.6, 37.2, 30.6, 26.8, 26.2, 25.8; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C18H25NNaO4+ 342.1676; Found 342.1680.

To a solution of (bromomethyl)cyclohexane (91 mg, 0.51 mmol, 1.0 equiv.) in DMF (1 mL) was added 2,4-DM-BENAC-K (1b) (154 mg, 0.53 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 19.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 8b (149 mg, 84%) as a colorless oil. IR (film) 2925, 2851, 1732, 1205 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.25 (1H, m), 6.48–6.46 (2H, m), 5.18 (2H, s), 3.83 (3H, s), 3.82 (3H, s), 3.56 (2H, d, J = 6.8 Hz), 2.49 (3H, s), 1.68–1.52 (6H, m), 1.17–1.07 (3H, m), 0.91–0.84 (2H, m); 13C{1H} NMR (125 MHz, CDCl3) δ: 173.2, 161.6, 159.1, 155.2, 131.7, 115.8, 103.8, 98.4, 64.1, 55.4, 55.3, 49.7, 37.1, 30.6, 26.7, 26.3, 25.9; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C19H27NNaO5+ 372.1781; Found 372.1782.

To a solution of benzyl bromide (120 µL, 171 mg, 1.00 mmol) in DMF (1 mL) was added PM-BENAC-K (1a) (288 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at room temperature for 0.8 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 9a (314 mg, quantative yield) as a colorless oil. IR (film) 2959, 1737, 1700, 1202 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.27–7.21 (3H, m), 7.20–7.18 (2H, m), 7.17 (2H, AA′XX′, JAA′ = 2.6 Hz, JXX′ = 2.6 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 6.85 (2H, AA′XX′, JAA′ = 2.6 Hz, JXX′ = 2.6 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.11 (2H, s), 4.91 (2H, s), 3.81 (3H, s), 2.55 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.9, 159.8, 154.4, 137.6, 130.2, 128.3, 127.8, 127.2, 126.9, 113.9, 68.5, 55.2, 46.9, 26.7; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C18H19NNaO4+ 336.1206; Found 336.1209.

To a solution of benzyl bromide (105 mg, 0.60 mmol) in DMF (3 mL) was added 2,4-DM-BENAC-K (1b) (179 mg, 0.64 mmol, 1.1 equiv.), and the reaction mixture was stirred at room temperature for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 9b (202 mg, 97%) as a colorless oil. IR (film) 2961, 1733, 1206 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.23–7.20 (5H, m), 7.13 (1H, d, J = 8.2 Hz), 6.44 (1H, d, J = 2.3 Hz), 6.42 (1H, dd, J = 8.2, 2.3 Hz), 5.17 (2H, s), 4.89 (2H, s), 3.81 (3H, s), 3.75 (3H, s), 2.53 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 173.1, 161.7, 159.1, 154.6, 137.8, 131.8, 128.19, 128.17, 127.1, 115.6, 103.9, 98.4, 64.3, 55.4, 55.3, 47.0, 26.7; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C19H21NNaO5+ 366.1312; Found 366.1315.

To a solution of allyl bromide (87 µL, 121 mg, 1.0 mmol) in DMF (1 mL) was added PM-BENAC-K (1a) (288 mg, 1.10 mmol, 1.1 equiv.), and the reaction mixture was stirred at room temperature for 0.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 10a (264 mg, quantative yield) as a colorless oil. IR (film) 2958, 1734, 1701, 1216 cm−1;1H-NMR (500 MHz, CDCl3) δ: 7.31 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 6.90 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 5.78 (1H, ddt, J = 17.6, 10.1, 5.7 Hz), 5.16 (2H, s), 5.090 (1H, dq, J = 10.1, 1.4 Hz), 5.089 (1H, dq, J = 17.6, 1.4 Hz), 4.33 (2H, dt, J = 5.7, 1.4 Hz), 3.82 (3H, s), 2.51 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.6, 159.9, 154.4, 132.9, 130.2, 127.1, 116.9, 114.0, 68.4, 55.3, 46.0, 26.7; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C14H17NNaO4+ 286.1050; Found 286.1050.

To a solution of allyl bromide (200 µL, 286 mg, 2.36 mmol, 1.14 mmol) in DMF (2.5 mL) was added 2,4-DM-BENAC-K (1b) (608 mg, 2.08 mmol, 1.0 equiv.), and the reaction mixture was stirred at room temperature for 5.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 10b (514 mg, 84%) as a colorless oil. IR (film) 3083, 2960, 1734, 1700, 1209 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.24 (1H, m), 6.48–6.45 (2H, m), 5.78 (1H, ddt, J = 17.3, 10.1, 5.8 Hz), 5.19 (2H, s), 5.09 (1H, dq, J = 17.3, 1.4 Hz), 5.07 (1H, dq, J = 10.1, 1.4 Hz), 4.30 (2H, dt, J = 5.8, 1.4 Hz), 3.819 (3H, s), 3.818 (3H, s), 2.50 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.6, 161.6, 159.1, 154.5, 133.0, 131.6, 116.9, 115.7, 103.9, 98.5, 64.3, 55.35, 55.30, 46.1, 26.6; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C15H19NNaO5+ 316.1155; Found 316.1158.

To a solution of propargyl bromide (38.0 µL, 68.4 mg, 0.50 mmol, 1.0 equiv.) in DMF (3 mL) was added PM-BENAC-K (1a) (144 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 0 °C for 1.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 11a (99 mg, 76%) as a colorless oil. IR (film) 3285, 2961, 1742, 1707, 1214 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.36 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 6.91 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 5.22 (2H, s), 4.50 (2H, d, J = 2.4 Hz), 3.82 (3H, s), 2.52 (3H, s), 2.15 (1H, t, J = 2.4 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 171.9, 159.9, 153.5, 130.2, 126.9, 114.0, 79.1, 70.6, 68.8, 55.2, 33.3, 26.4; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C14H15NNaO4+ 284.0893; Found 284.0894.

To a solution of propargyl bromide (200 µL, 316 mg, 2.66 mmol, 1.33 mmol) in DMF (2 mL) was added 2,4-DM-BENAC-K (1b) (608 mg, 2.08 mmol, 1.0 equiv.), and the reaction mixture was stirred at room temperature for 1.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 11b (454 mg, 69%) as a colorless solid. mp 63–64 °C; IR (film) 3284, 2962, 2837, 1738, 1208 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.29 (1H, m), 6.48–6.46 (2H, m), 5.25 (2H, s), 4.48 (2H, d, J = 2.4 Hz), 3.83 (3H, s), 3.82 (3H, s), 2.50 (3H, s), 2.13 (1H, t, J = 2.4 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.1, 161.7, 159.1, 153.7, 131.6, 115.6, 103.9, 98.5, 79.3, 70.4, 64.7, 55.4 (×2), 33.4, 26.5; HRMS (ESI/Orbitrap) m/z: [M + Na]− Calcd for C15H17NNaO5+ 314.0999; Found 314.1001.

To a solution of ethyl bromoacetate (172 mg, 1.03 mmol) in DMF (5.5 mL) was added PM-BENAC-K (1a) (289 mg, 1.11 mmol, 1.1 equiv.), and the reaction mixture was stirred at room temperature for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (15% EtOAc in n-hexane) afforded 12a (305 mg, 96%) as a colorless oil. IR (film) 2966, 1746, 1705, 1202 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 7.28 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.6 Hz, JAX′ = 0 Hz), 6.89 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.6 Hz, JAX′ = 0 Hz), 5.16 (2H, s), 4.47 (2H, s), 4.13 (2H, q, J = 7.4 Hz), 3.81 (3H, s), 2.57 (3H, s), 1.20 (3H, t, J = 7.4 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 172.5, 168.6, 159.9, 153.6, 130.2, 126.8, 114.0, 68.7, 61.3, 55.2, 44.9, 26.2, 14.0; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C15H19NNaO6+ 332.1105; Found 332.1106.

To a solution of ethyl bromoacetate (55.3 µL, 83.5 mg, 0.50 mmol) in DMF (3 mL) was added 2,4-DM-BENAC-K (1b) (162 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 0 °C for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 12b (131 mg, 77%) as a colorless oil; IR (film) 2967, 1742, 1200 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.21 (1H, m), 6.46–6.44 (2H, m), 5.20 (2H, s), 4.45 (2H, m), 4.12 (2H, q, J = 7.2 Hz), 3.812 (3H, s), 3.809 (3H, s), 2.54 (3H, s), 1.20 (3H, t, J = 7.2 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 172.5, 168.5, 161.6, 159.0, 153.7, 131.5, 115.4, 103.9, 98.4, 64.6, 61.1, 55.3, 55.2, 45.0, 26.1, 13.9; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C16H21NNaO7+ 362.1210; Found 362.1213.

To a solution of methyl 2-bromopropionate (167 mg, 1.00 mmol, 1.0 equiv.) in toluene (5.5 mL) was added 18-crown-6 (271 mg, 1.0 mmol, 1.0 equiv.) and PM-BENAC-K (1a) (286 mg, 1.1 mmol, 1.0 equiv.), and the reaction mixture was stirred at 60 °C for 7.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (25% EtOAc in n-hexane) afforded 13a (222 mg, 72%) as a colorless oil. IR (film) 2951, 1744, 1703, 1247 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 7.30 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 6.90 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 5.29 (1H, q, J = 6.9 Hz), 5.20 (1H, d, J = 11.7 Hz), 5.12 (1H, d, J = 11.7 Hz), 3.82 (3H, s), 3.53 (3H, s), 2.52 (3H, s), 1.45 (3H, d, J = 6.9 Hz); 13C{1H} NMR (125 MHz, CDCl3) δ: 172.2, 171.1, 160.0, 153.6, 130.5, 126.6, 114.1, 68.7, 55.3, 52.2, 51.7, 26.5, 15.4; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C15H19NNaO6+ 332.1105; Found 332.1106.

To a solution of methyl 2-bromopropionate (83.4 mg, 0.50 mmol, 1.0 equiv.) in CH3CN (3 mL) was added 18-crown-6 (206 mg, 0.78 mmol, 1.5 equiv.) and 2,4-DM-BENAC-K (1b) (161 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at room temperature for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc and 0.5% Et3N in n-hexane) afforded 13b (70.2 mg, 46%) as a colorless oil. IR (film) 2950, 1741 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.22 (1H, m), 6.47–6.45 (2H, m), 5.26 (1H, q, J = 6.8 Hz), 5.20 (1H, d, J = 11.9 Hz), 5.19 (1H, d, J = 11.9 Hz), 3.825 (3H, s), 3.816 (3H, s), 3.54 (3H, s), 2.50 (3H, s), 1.44 (3H, d, J = 6.8 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 172.3, 171.1, 161.8, 159.2, 153.7, 131.8, 115.3, 103.9, 98.4, 64.6, 55.32, 55.27, 52.0, 51.8, 26.4, 15.2; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C16H21NNaO7+ 362.1210; Found 362.1208.

To a solution of 5-chloro-1-pentyne (103 mg, 1.00 mmol, 1.0 equiv.) in DMF (5.5 mL) was added PM-BENAC-K (1a) (287 mg, 1.1 mmol, 1.0 equiv.), and the reaction mixture was stirred at 60 °C for 46.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (15% EtOAc in n-hexane) afforded 14a (267 mg, 92%) as a colorless solid. mp 45–48 °C; IR (film) 3289, 2960, 1734, 1698, 1172 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 7.34 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 6.91 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 5.17 (2H, s), 3.82 (3H, s), 3.82 (2H, t, J = 7.3 Hz), 2.49 (3H, s), 2.17 (2H, td, J = 7.1, 2.7 Hz), 1.91 (1H, t, J = 2.7 Hz), 1.74 (2H, tt, J = 7.3, 7.1 Hz); 13C{1H} NMR (125 MHz, CDCl3) δ: 172.8, 159.9, 154.4, 130.2, 127.1, 114.0, 83.3, 68.6, 68.3, 55.2, 43.3, 27.3, 26.8, 16.1; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C16H19NNaO4+ 312.1206; Found 312.1204.

To a solution of 5-chloro-1-pentyne (52 µL, 51 mg, 0.50 mmol, 1.0 equiv.) in DMF (1 mL) was added 2,4-DM-BENAC-K (1b) (161 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 18.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 14b (94.3 mg, 59%) as a colorless solid of mp 49–51 °C. IR (film) 3288, 2961, 1732 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 7.26 (1H, d, J = 8.7 Hz), 6.47 (1H, d, J = 2.3 Hz), 6.46 (1H, dd, J = 8.7, 2.3 Hz), 5.19 (2H, s), 3.83 (3H, s), 3.82 (3H, s), 3.78 (2H, t, J = 7.3 Hz), 2.48 (3H, s), 2.15 (1H, td, J = 7.2, 2.7 Hz), 1.87 (1H, t, J = 2.7 Hz), 1.74 (2H, quintet, J = 7.2 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 172.9, 161.6, 159.1, 154.5, 131.7, 115.6, 103.8, 98.4, 83.4, 68.4, 64.3, 55.30, 55.27, 43.2, 27.3, 26.7, 16.1; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C17H21NNaO5+ 342.1312; Found 342.1312.
5a from Benzyl 3-Iodopropyl EtherTo a solution of benzyl 3-iodopropyl ether (140 mg, 0.50 mmol) in DMF (3 mL) was added PM-BENAC-K (1a) (144 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 5a (183 mg, 98%) as a colorless oil.
5b from Benzyl 3-Iodopropyl EtherTo a solution of benzyl 3-iodopropyl ether (139 mg, 0.50 mmol) in DMF (3 mL) was added 2,4-DM-BENAC-K (1b) (162 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 2.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (20% EtOAc in n-hexane) afforded 5b (119 mg, 60%) as a colorless oil.
5a from 3-Benzyloxypropyl p-ToluenesulfonateTo a solution of 3-benzyloxypropyl p-toluenesulfonate (160 mg, 0.50 mmol) in DMF (3 mL) was added PM-BENAC-K (1a) (145 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 1.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (50% Et2O in n-hexane) afforded 5a (178 mg, 96%) as a colorless oil.
5b from 3-Benzyloxypropyl p-ToluenesulfonateTo a solution of 3-benzyloxypropyl p-toluenesulfonate (161 mg, 0.50 mmol) in DMF (3 mL) was added 2,4-DM-BENAC-K (1b) (162 mg, 0.55 mmol, 1.1 equiv.), and the reaction mixture was stirred at 60 °C for 3.0 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (50% Et2O in n-hexane) afforded 5b (159 mg, 80%) as a colorless oil.

To a solution of ((2R,3S)-3-((tert-butyldimethylsilyl)oxy)tetrahydro-2H-pyran-2-yl)methyl trifluoromethanesulfonate42) (114 mg, 0.30 mmol, 1.0 equiv.) in toluene (0.3 mL) was added PM-BENAC-K (1a) (87 mg, 0.33 mmol, 1.1 equiv.) and 18-crown-6 (80.0 mg, 0.30 mmol, 1.0 equiv.) and the reaction mixture was stirred at room temperature for 0.8 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 15a (120.4 mg, 89%) as a colorless oil. [α]23D +45.5 (c 1.27, CHCl3); IR (film) 2953, 2936, 2856, 1738, 1703, 1224 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.35 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 6.89 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.18 (1H, d, J = 11.9 Hz), 5.16 (1H, d, J = 11.9 Hz), 4.05 (1H, dd, J = 13.9, 2.8 Hz), 3.98 (1H, dd, J = 13.9, 9.9 Hz), 3.82 (3H, s), 3.77 (1H, ddt, J = 11.3, 4.5, 1.7 Hz), 3.33 (1H, ddd, J = 10.3, 8.9, 4.7 Hz), 3.22 (1H, ddd, J = 9.8, 8.9, 3.1 Hz), 3.11 (1H, td, J = 11.6, 2.8 Hz), 2.48 (3H, s), 1.97 (1H, ddtd, J = 12.8, 4.7, 3.3, 1.7 Hz), 1.63 (1H, ddddd, J = 13.2, 12.8, 11.9, 4.8, 3.3 Hz), 1.56 (1H, ddddd, J = 13.2, 4.8, 3.3, 2.8, 1.7 Hz), 1.39 (1H, tdd, J = 12.8, 10.4, 4.8 Hz), 0.87 (9H, s), 0.053 (3H, s), 0.046 (3H, s); 13C{1H} NMR (125 MHz, CDCl3) δ: 173.0, 159.7, 155.0, 130.2, 127.5, 113.9, 80.3, 70.4, 68.1, 67.5, 55.3, 45.7, 33.3, 26.6, 25.7, 25.3, 17.8, −3.9, −4.9; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C23H37NNaO7Si+ 474.2282; Found 474.2289.

To a solution of ((2R,3S)-3-((tert-butyldimethylsilyl)oxy)tetrahydro-2H-pyran-2-yl)methyl trifluoromethanesulfonate42) (114 mg, 0.30 mmol, 1.0 equiv.) in toluene (0.3 mL) was added 2,4-DM-BENAC-K (1b) (98 mg, 0.33 mmol, 1.1 equiv.) and 18-crown-6 (80.0 mg, 0.30 mmol, 1.0 equiv.) and the reaction mixture was stirred at room temperature for 2.5 h. The reaction was quenched with aqueous saturated NH4Cl solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 15b (131 mg, 90%) as a colorless oil of [α]23D +32.1 (c 1.02, CHCl3). IR (film) 2953, 2856, 1736, 1210 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 7.26 (1H, ABX, νAB = 1.5 Hz, JAB = 2.3 Hz, JAX = 8.4 Hz, JBX = 0 Hz), 6.46 (2H, ABX, νAB = 1.5 Hz, JAB = 2.3 Hz, JAX = 8.4 Hz, JBX = 0 Hz), 5.23 (1H, d, J = 11.9 Hz), 5.18 (1H, d, J = 11.9 Hz), 4.00 (2H, ABX, νAB = 3.0 Hz, JAB = 13.9 Hz, JAX = 2.8 Hz, JBX = 9.9 Hz), 3.83 (3H, s), 3.81 (3H, s), 3.78 (1H, ddt, J = 11.3, 4.5, 1.7 Hz), 3.33 (1H, ddd, J = 10.2, 8.8, 4.5 Hz), 3.27 (1H, ABX-d, νAB = 3.0 Hz, JAB = 13.9 Hz, JAX = 2.8 Hz, JBX = 9.9 Hz, Jdoublet = 8.8 Hz), 3.13 (1H, td, J = 11.6, 2.8 Hz), 2.47 (3H, s), 1.95 (1H, ddtd, J = 12.8, 4.7, 3.3, 1.7 Hz), 1.63 (1H, ddddd, J = 13.2, 12.8, 11.9, 4.8, 3.3 Hz), 1.56 (1H, ddddd, J = 13.2, 4.8, 3.3, 2.8, 1.7 Hz), 1.38 (1H, tdd, J = 12.8, 10.4, 4.8 Hz), 0.83 (9H, s), 0.03 (3H, s), 0.02 (3H, s); 13C{1H} NMR (125 MHz, CDCl3) δ: 173.2, 161.4, 158.9, 155.1, 131.5, 116.1, 103.9, 98.4, 80.1, 70.4, 67.5, 64.0, 55.4 (x2), 45.8, 33.4, 26.6, 25.6, 25.3, 17.8, –3.9, –4.9; HRMS (ESI/Orbitrap) m/z: [M + Na]+ Calcd for C24H39NNaO7Si+ 504.2388; Found 504.2388.

A solution of benzyl acetyl(3-benzyloxypropyl)carbamate (16)18) (17 mg, 0.050 mmol) in CH2Cl2/CF3CO2H (1 : 1) (0.5 mL) was stirred at room temperature for 26 h. The reaction was quenched with saturated aqueous NaHCO3 solution and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (90% EtOAc in n-hexane → EtOAc) afforded N-(3-(benzyloxy)propyl)acetamide (17) (8.6 mg, 83%) as a colorless oil. IR (film) 3295, 2930, 2861, 1651, 1556 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.37–7.28 (5H, m), 6.15 (1H, br s), 4.50 (2H, s), 3.58 (2H, t, J = 5.8 Hz), 3.36 (2H, q, J = 6.1 Hz), 1.88 (3H, s), 1.80 (2H, quintet, J = 6.0 Hz), 13C{1H} NMR (150 MHz, CDCl3) δ: 170.0, 138.1, 128.4, 127.7, 127.6, 73.1, 69.1, 38.1, 28.9, 23.2; HRMS (DART/Orbitrap) m/z: [M + H]+ Calcd for C12H18NO2+ 208.1332; Found 208.1330.
Formic Acid-Mediated Deprotection of a Moz Group of 5aA solution of 5a (20 mg, 0.050 mmol) in CH2Cl2/HCO2H (1 : 1) (0.5 mL) was stirred at room temperature for 1 h. The reaction was quenched with saturated aqueous NaHCO3 solution and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (90% EtOAc in n-hexane → EtOAc) afforded 17 (10.8 mg, quantative yield) as a colorless oil.
Formic Acid-Mediated Deprotection of a Dmoz Group of 5bA solution of 5b (20 mg, 0.050 mmol) in CH2Cl2/CH3CO2H (1 : 1) (0.5 mL) was stirred at room temperature for 24 h. The reaction was quenched with saturated aqueous NaHCO3 solution and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Flash chromatography on silica gel (EtOAc) afforded 17 (10.1 mg, 96%) as a colorless oil.
TsOH·H2O-Catalyzed Deprotection of a Dmoz Group of 5bTo a solution of 5b (20 mg, 0.050 mmol) in CH2Cl2/CH3OH (1 : 1) (1.0 mL) was added p-TsOH·H2O (2.0 mg, 0.01 mmol, 0.2 equiv.), and the reaction mixture was stirred at room temperature for 24 h. The reaction was quenched with Et3N and the resulting mixture was concentrated under reduced pressure. Flash chromatography on silica gel (EtOAc) afforded 17 (11.0 mg, quantative yield) as a colorless oil.
CAN-Mediated Deprotection of a Dmoz Group of 5bTo a solution of 5b (84 mg, 0.21 mmol) in CH3CN (2.1 mL) and H2O (0.21 mL) was added ceric ammonium nitrate (233 mg, 2.0 equiv.), and the reaction mixture was stirred at room temperature for 2.5 h. The reaction was quenched with saturated aqueous NaHCO3 and saturated aqueous Na2S2O3 solution, and the resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over MgSO4, filtrated, and concentrated under reduced pressure. Flash chromatography on silica gel (EtOAc) afforded 17 (30.7 mg, 71%) as a colorless oil.
Hydrogenation of 5aTo a solution of 5a (53.5 mg, 0.14 mmol) in EtOAc (1 mL) was added 5% Pd/C (7 mg), and the reaction mixture was stirred under hydrogen at room temperature for 3 h. After replacing Ar atmosphere, additional 10% Pd/C (4.8 mg) was added, and then the mixture was stirred under hydrogen at room temperature for additional 1 h. The reaction mixture was filtered through a celite pad and the filtrate was concentrated under reduced pressure. Flash chromatography on silica gel (EtOAc) afforded 17 (19.6 mg, 68%) as a colorless oil.

To a solution of 12a (88 mg, 0.28 mmol, 1.0 equiv.) in MeOH (1.0 mL) was added K2CO3 (3.8 mg, 0.027 mmol, 0.1 equiv.), and the reaction mixture was stirred at 0 °C for 1.5 h. The reaction was quenched with AcOH, and the resulting mixture was filtrated and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 18a (60 mg, 84%) as a colorless oil. IR (film) 3360, 2955, 2839, 1752, 1721, 1515 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.30 (2H, br d, J = 8.6 Hz), 6.87 (2H, AA′XX′, JAA′ = 2.5 Hz, JXX′ = 2.5 Hz, JAX = 8.8 Hz, JAX′ = 0 Hz), 5.26 (1H, br s), 5.06 (2H, s), 3.97 (2H, d, J = 5.5 Hz), 3.80 (3H, s), 3.75 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 170.4, 159.6, 156.3, 130.0, 128.3, 113.9, 66.9, 55.2, 52.3, 42.6; HRMS (DART/Orbitrap) m/z: [M + NH4]+ Calcd for C12H19N2O5+ 271.1288; Found 271.1290

To a solution of 12b (129 mg, 0.38 mmol, 1.0 equiv.) in MeOH (1.5 mL) was added K2CO3 (5.3 mg, 0.038 mmol, 0.1 equiv.), and the reaction mixture was stirred at 0 °C for 1.5 h. The reaction was quenched with AcOH, and the resulting mixture was filtrated and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 18b (86 mg, 80%) as a colorless solid of mp 75–77 °C. IR (film) 3373, 2953, 1752, 1719, 1509, 1207 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.25 (1H, m), 6.47–6.45 (2H, m), 5.21 (1H, br s), 5.11 (2H, s), 3.98 (2H, d, J = 5.5 Hz), 3.82 (3H, s), 3.81 (3H, s), 3.75 (3H, s); 13C{1H} NMR (150 MHz, CDCl3) δ: 170.5, 161.3, 158.9, 156.5, 131.4, 116.9, 104.0, 98.5, 62.5, 55.5, 55.4, 52.3, 42.6; HRMS (DART/Orbitrap) m/z: [M–H]– Calcd for C13H16NO6+ 282.0983; Found 282.0981.

To a solution of 13a (77 mg, 0.25 mmol, 1.0 equiv.) in MeOH (1 mL) was added K2CO3 (3.2 mg, 0.022 mmol, 0.1 equiv.), and the reaction mixture was stirred at 0 °C for 1 h. The reaction was quenched with AcOH, and the resulting mixture was filtrated and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 19a (62 mg, 89%) as a colorless solid of mp 58–62 °C. IR (film) 3345, 2954, 2838, 1715, 1515 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 7.29 (2H, br d, J = 8.2 Hz), 6.88 (2H, AA′XX′, JAA′ = 2.4 Hz, JXX′ = 2.4 Hz, JAX = 8.7 Hz, JAX′ = 0 Hz), 5.33 (1H, br d, J = 6.0 Hz), 5.05 (1H, d, J = 11.9 Hz), 5.02 (1H, d, J = 11.9 Hz), 4.38 (1H, quintet, J = 7.3 Hz), 3.80 (3H, s), 3.73 (3H, s), 1.40 (3H, d, J = 7.3 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ: 173.4, 159.5, 155.6, 129.9, 128.3, 113.9, 66.5, 55.2, 52.4, 49.3, 18.5. HRMS (DART/Orbitrap) m/z: [M + NH4]+ Calcd for C13H21N2O5+ 285.1449; Found 285.1449.
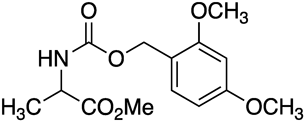
To a solution of 13b (40 mg, 0.12 mmol, 1.0 equiv.) in MeOH (0.8 mL) was added K2CO3 (1.2 mg, 0.012 mmol, 0.1 equiv.), and the reaction mixture was stirred at 0 °C for 4.5 h. The reaction was quenched with AcOH, and the resulting mixture was filtrated and concentrated under reduced pressure. Flash chromatography on silica gel (30% EtOAc in n-hexane) afforded 19b (18 mg, 50%) as a colorless solid of mp 100–102 °C. IR (film) 3352, 2954, 2839, 1715, 1510, 1208 cm−1; 1H-NMR (600 MHz, CDCl3) δ: 7.26 (1H, m), 6.47–6.45 (2H, m), 5.27 (1H, br d, J = 6.1 Hz), 5.10 (1H, d, J = 11.6 Hz), 5.08 (1H, d, J = 11.6 Hz), 4.39 (1H, quintet, J = 7.0 Hz), 3.82 (3H, s), 3.81 (3H, s), 3.74 (3H, s), 1.40 (3H, d, J = 7.2 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ: 173.5, 161.2, 158.9, 155.8, 131.4, 117.0, 104.0, 98.5, 62.2, 55.45, 55.36, 52.4, 49.5, 18.7; HRMS (DART/Orbitrap) m/z: [M–H]− Calcd for C14H18NO6+ 296.1140; Found 296.1137.
This research was partially supported by Grant-in-Aids for Scientific Research (C) (20K05503 and 22K06539) from the Japan Society for the Promotion of Science (JSPS), and research fund from the Research Institute of Meijo University.
The authors declare no conflict of interest.
This article contains supplementary materials.