Experimental
General Experimental ProceduresAll reactions were monitored by TLC using Merck 60 F254 precoated silica gel plates (0.25 mm thick) and Fuji Silysia Chemical precoated amino-silica gel plates (0.25 mm thick). UV spectra were recorded in MeOH on a JASCO V-560 instrument. Specific optical rotations were measured using a JASCO P-2200 polarimeter. Circular dichroism spectra were recorded on a JASCO J-1100 spectrometer. Fourier transform (FT) IR spectra were recorded on a JASCO FT/IR-4700. 1H- and 13C-NMR spectra were recorded on a JEOL ECZ 400 (400 MHz 1H-NMR, 100 MHz for 13C-NMR), ECS 400 (400 MHz 1H-NMR, 100 MHz for 13C-NMR), JEOL ECX 500 (500 MHz 1H-NMR, 125 MHz for 13C-NMR), and ECZ 600 (600 MHz for 1H-NMR, 150 MHz for 13C-NMR) FT-NMR spectrometer instrument. Data for 1H-NMR are reported as chemical shifts (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, dd = double doublet, ddd = double double doublet, dt = double triplet, q = quartet, m = multiplet, br = broad), coupling constant (Hz), integration, and assignment. Data for 13C-NMR are reported as chemical shifts. The high-resolution mass spectra were recorded on a JEOL AccuTOF LC-plus JMS-T100LP. Flash chromatography was performed using Kanto Chemical silica gel 60N and Fuji Silysia Chemical amino-silica gel (SiO2-NH, NH-DM2035). HPLC analysis was performed on a JASCO MD-4017; UV detection was monitored at an appropriate wavelength using Daicel Chiralcel OD-H (0.46 × 25 cm).
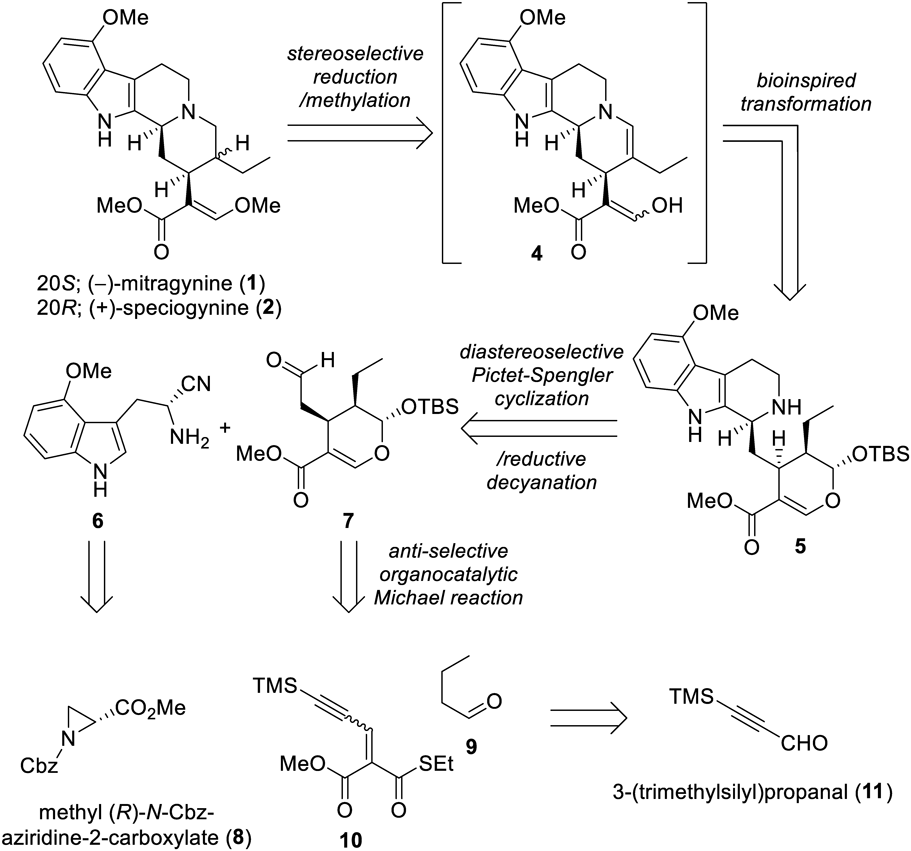
Preparation of Compound 14To a solution of known aziridine 8 (1.0 g, 4.25 mmol) and 4-methoxyindole (12, 1.25 g, 8.50 mmol) in CH2Cl2 (17 mL) was added Yb(OTf)3 (2.64 g, 4.25 mmol) under Ar atmosphere. The reaction mixture was stirred for 24 h at r.t. under Ar atmosphere in the dark. To the resulting mixture was added additional Yb(OTf)3 (1.32 g, 2.13 mmol), followed by stirring for a further 48 h at r.t. The mixture was then quenched with saturated aqueous NaHCO3 and the resulting mixture was filtered through a Celite pad, eluted with CHCl3. The filtrate was extracted three times with CHCl3. The combined organic layer was dried over Na2SO4 and then concentrated under reduced pressure. Flash chromatography (SiO2, 22–28% AcOEt/n-hexane gradient) provided an inseparable regioisomer mixture of 13 (780 mg, r.r. = 6.7 : 1). To a solution of a mixture of 13 in aqueous THF (34 mL, THF/H2O = 10 : 1) was added 2 M aqueous LiOH (3.06 mL, 6.12 mmol) at r.t. under Ar atmosphere. The reaction mixture was stirred for 2.5 h at r.t. before quenching with 2 M aqueous HCl (pH 2). Brine was added to the resulting mixture and the aqueous layer was extracted three times with AcOEt. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. Obtained crude materials were dissolved in anhydrous THF (40.8 mL), then ethyl chloroformate (272 µL, 2.86 mmol) and triethylamine (853 µL, 6.12 mmol) were added at 0 °C under Ar atmosphere. The reaction mixture was stirred for 30 min at 0 °C. To the resulting mixture was added 1.0 M aqueous NH4Cl (3.06 mL, 3.06 mmol) at 0 °C. The reaction mixture was stirred for an additional 20 min at 0 °C. It was then quenched with saturated aqueous NaHCO3 and the aqueous layer was extracted three times with AcOEt. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 2%–5% MeOH/CHCl3 gradient) afforded amide 14 (607.5 mg, 39% over three steps).
Compound 14: White amorphous powder; [α]23D +2.6 (c 0.31, MeOH); IR (ATR) νmax cm−1 3448, 3338, 2945, 2480, 1674, 1620, 1589, 1525, 1502, 1423, 1356, 1313, 1257, 1153, 1095, 1039, 966, 910, 860, 775, 752, 735, 719, 694, 617; high resolution (HR)MS (electrospray ionization (ESI)) [M + Na]+ Calcd. for [C20H21N3Na1O4]+: 390.1430, Found: 390.1435; UV (MeOH) λmax 291, 281, 265, 220 nm; 1H-NMR (400 MHz, (CD3)2SO) δ: 10.8 (br s, 1H), 7.34–7.19 (m, 5H), 7.03 (br s, 1H), 6.96–6.92 (m, 3H), 6.44 (d, J = 6.4 Hz, 1H), 4.95 (s, 2H), 4.23 (dt, J = 8.8, 4.4 Hz, 1H), 3.84 (s, 3H), 3.28 (dt, J = 14.4, 3.6 Hz, 1H), 2.98 (ddd, J = 14.4, 10.0, 3.2 Hz, 1H); 13C-NMR (150 MHz, (CD3)2SO) δ: 174.2, 155.8, 154.1, 137.8, 137.1, 128.4, 127.7, 127.4, 122.2, 121.8, 117.0, 110.6, 105.0, 98.9, 65.2, 56.2, 55.1, 39.9, 39.8, 39.7, 39.5, 39.4, 39.2, 39.1, 29.3.
Preparation of (R)-4-Methoxy-α-cyanotryptamine (6)To a solution of amide 14 (608 mg, 1.65 mmol) in anhydrous pyridine (11 mL) was added phosphoryl chloride (355 µL, 3.80 mmol) at 0 °C under Ar atmosphere. The reaction mixture was stirred for 50 min at 0 °C. MeOH was added at 0 °C to the resulting mixture and stirred for 10 min, followed by concentration under reduced pressure. The resulting mixture was dissolved in Et2O, and the organic layer was washed with 1 M aqueous NaOH three times, and with brine, dried over Na2SO4, and then concentrated under reduced pressure. Flash chromatography (SiO2, 1% MeOH/CHCl3) gave a dehydrated compound (510 mg, 88%). To a solution of the dehydrated compound (500 mg, 1.43 mmol) in degassed 1,4-dioxane (10.4 mL) was added 10% Pd/C (152 mg, 0.143 mmol) at r.t. under Ar atmosphere. The resulting mixture was purged with a stream of hydrogen and the reaction mixture was stirred for 13 h at r.t. under H2 atmosphere. The resulting mixture was filtered with a Celite pad with CHCl3. The filtrate was concentrated under reduced pressure and the resulting residue was purified by flash chromatography (SiO2, 60% AcOEt/n-hexane, then 5% MeOH/CHCl3) to afford (R)-4-methoxy-α-cyanotryptamine (6, 260.0 mg, 84%).
(R)-4-Methoxy-α-cyanotryptamine (6): Pale pink amorphous powder; [α]23D –15.0 (c 1.96, CHCl3); IR (ATR) νmax cm−1 3363, 3194, 2914, 1741, 1587, 1547, 1510, 1431, 1356, 1257, 1167, 1132, 1086, 1053, 955, 895, 852, 814, 683, 623; HRMS (ESI) [M–CN]+ Calcd. for [C11H13N2O1]+: 189.1028, Found: 189.1025; UV (MeOH) λmax 290, 281, 267, 220 nm; 1H-NMR (600 MHz, CDCl3) δ: 8.22 (br s, 1H), 7.11 (t, J = 7.8 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 6.52 (d, J = 7.8 Hz, 1H), 4.18 (t, J = 7.2 Hz, 1H), 3.93 (s, 3H), 3.33 (dd, J = 13.8, 7.2 Hz, 1H), 3.27 (dd, J = 13.8, 7.2 Hz, 1H); 13C-NMR (150 MHz, CDCl3) δ: 154.4, 138.2, 123.3, 122.6, 122.5, 117.0, 110.0, 104.9, 99.8, 55.3, 45.4, 33.6.
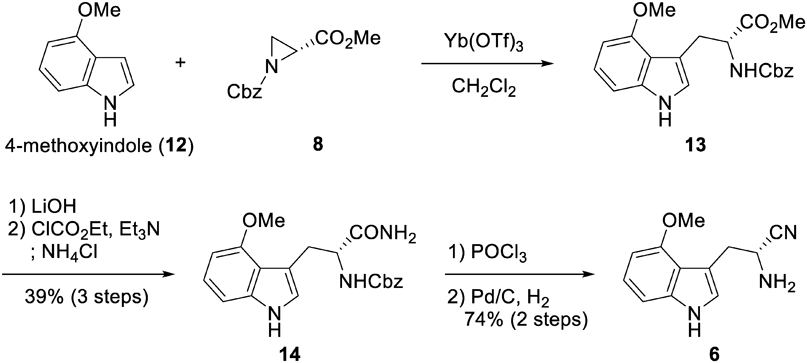
Preparation of Compound 17To the solution of an E and Z mixture of half thioester malonate derivative 10 (500 mg, 1.85 mmol) and diphenylprolinol trimethyl silyl ether (18.0 mg, 0.0555 mmol) in Et2O (9.2 mL) was added n-butyraldehyde (9, 167 µL, 1.85 mmol) at 0 °C under Ar atmosphere. The reaction mixture was stirred for 40 h at 0 °C. The resulting mixture was concentrated under reduced pressure. Crude 1H-NMR revealed that anti-adduct 16 was the main product (diastereomer ratio of 16; anti:syn = 11 : 1). The crude materials of 16 were solved to degassed THF (3.7 mL), then 10% Pd/C (197 mg, 0.185 mmol) and Et3SiH (886 µL, 5.55 mmol) were added at 0 °C under Ar atmosphere. The reaction mixture was stirred for 4.5 h at r.t, whereafter an additional 10% Pd/C (98.4 mg, 0.0924 mmol) and Et3SiH (443 µL, 2.77 mmol) were added at r.t. under Ar atmosphere. This was stirred for 5 h at r.t. before the addition of 1 M aqueous HCl (3.7 mL). The resulting mixture was stirred for a further 18 h at r.t. before filtration with a Celite pad eluted with AcOEt, and partitioned between an AcOEt layer and an aqueous layer. The aqueous layer was extracted three times with AcOEt. The combined organic layer was dried over MgSO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 5%–20% AcOEt/n-hexane gradient) afforded two diastereomers of dihydropyran 17 (444.5 mg, 85% over two steps, d.r. = 4 : 1).
Compound 17: Yellow waxy solid; IR (ATR) νmax cm−1 3370, 2953, 1672, 1629, 1444, 1383, 1311, 1247, 1204, 1148, 1125, 1100, 1030, 1015, 981, 945, 908, 879, 835; HRMS (ESI) [M + Na]+ Calcd. for [C14H22Na1O4Si1]+: 305.1185, Found: 305.1145; 1H-NMR (500 MHz, CDCl3) δ: 7.53 (s, 1H), 7.46 (s, 1H), 5.36 (dd, J = 12.0, 1.5 Hz, 1H), 5.22 (t, J = 7.5 Hz, 1H), 4.85 (d, J = 7.5 Hz, 1H), 3.77 (s, 3H), 3.75 (s, 3H), 3.58 (d, J = 5.0 Hz, 1H), 3.55 (d, J = 5.0 Hz, 1H), 3.34 (d, J = 7.5 Hz, 1H), 1.85–1.71 (m, 3H), 1.64–1.53 (m, 3H), 1.05 (t, J = 7.0 Hz, 3H), 1.02 (t, J = 7.0 Hz, 3H), 0.14 (s, 9H), 0.12 (s, 9H); 13C-NMR (125 MHz, CDCl3) δ: 166.9, 166.7, 153.3, 152.5, 107.2, 106.3, 104.0, 103.7, 97.1, 96.3, 90.1, 97.9, 51.7, 51.6, 42.2, 39.9, 26.8, 23.6, 21.6, 20.4, 11.4, 11.0, 0.17 (3C), 0.12 (3C).
Preparation of Compound 18To a solution of two diastereomers of dihydropyran 17 (1.0 g, 3.54 mmol) in dry DMF (11.8 mL) were added AgNO3 (782 mg, 4.60 mmol) and TBSCl (800.5 mg, 5.31 mmol) at 0 °C under Ar atmosphere. The reaction mixture was stirred for 1 h at r.t. under Ar atmosphere. Additional AgNO3 (391 mg, 2.30 mmol) and TBSCl (400 mg, 2.66 mmol) were then added under Ar atmosphere. The reaction mixture was stirred for 2.5 h at r.t. Additional AgNO3 (391 mg, 2.30 mmol) was then added under Ar atmosphere and the mixture was stirred for 15 min at r.t. The resulting mixture was directly filtered through a short plug of silica gel eluted with 20% AcOEt/n-hexane and the filtrate was added to saturated NaHCO3. The aqueous layer was extracted three times with AcOEt. The combined organic layer was washed four times each with water and brine, dried over MgSO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 1% AcOEt/n-hexane) afforded 18 (1.0 g, 87%). Enantiomeric excess was determined to be over 99% by HPLC (Chiralcel OD-H column) (see Supporting Information). Results for compound 18: 1.5% i-PrOH/n-hexane, 0.500 mL/min; major enantiomer tR = 7.27 min, minor enantiomer tR = 8.30 min.
Compound 18: Colorless oil; [α]22D –235.6 (c 0.71, CHCl3); IR (ATR) νmax cm−1 3315, 3269, 2954, 2931, 2883, 2858, 1714, 1639, 1462, 1437, 1392, 1298, 1254, 1228, 1167, 1130, 1088, 1001, 949, 924, 879, 837, 781, 764, 679, 663, 638, 617; HRMS (ESI) [M + Na]+ Calcd. for [C17H28Na1O4Si1]+: 347.1655, Found: 347.1669; 1H-NMR (400 MHz, CDCl3) δ: 7.48 (s, 1H), 5.18 (d, J = 8.4 Hz, 1H), 3.75 (s, 3H), 3.56 (dd, J = 5.2, 2.4 Hz, 1H), 2.15 (d, J = 2.4 Hz, 1H), 1.78 (m, 1H), 1.59 (m, 1H), 1.47 (m, 1H), 1.00 (t, J = 7.2 Hz, 3H), 0.93 (s, 9H), 0.15 (s, 3H), 0.15 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 167.1, 153.9, 106.6, 97.6, 82.4, 71.4, 51.6, 42.5, 25.8, 20.3, 18.1, 11.1, –4.1, –5.1.
Preparation of Secologanin Derivative 7To a solution of alkyne 18 (1.0 g, 3.1 mmol) in anhydrous THF (10.3 mL) was added 9-BBN (1.0 M in THF solution, 7.4 mL, 3.70 mmol) under Ar atmosphere. The reaction mixture was stirred for 2.5 h at r.t. The resulting mixture was quenched with H2O (1.0 mL) and then stirred for an additional 10 min. To the resulting mixture were added 30% aqueous H2O2 (1.57 mL, 15.4 mmol) and 1.0 M aqueous NaOH (3.08 mL, 3.08 mmol) at 0 °C. This reaction mixture was stirred for 80 min at r.t. under Ar atmosphere. After the addition of saturated aqueous NH4Cl, the aqueous layer was extracted three times with AcOEt. The combined organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. Flash chromatography, repeated three times (SiO2, 5%–50% AcOEt/n-hexane gradient then 10% MeOH/CHCl3) afforded secologanin derivative 7 (887 mg, 84%).
Secologanin Derivative 7: Colorless oil; [α]23D –141.1 (c 1.89, CHCl3); IR (ATR) νmax cm−1 2954, 2931, 2887, 2858, 1703, 1633, 1466, 1439, 1385, 1362, 1309, 1281, 1254, 1167, 1138, 1101, 1080, 1007, 918, 874, 837, 781, 675, 640; HRMS (ESI) [M + Na]+ Calcd. for [C17H30Na1O5Si1]+: 365.1760, Found: 365.1769; 1H-NMR (400 MHz, CDCl3) δ: 9.76 (dd, J = 3.2, 2.0 Hz, 1H), 7.49 (s, 1H), 5.06 (d, J = 7.6 Hz, 1H), 3.69 (s, 3H), 3.30 (dt, J = 7.6, 5.2 Hz, 1H), 2.48 (ddd, J = 15.2, 5.2, 2.0 Hz, 1H), 2.35 (ddd, J = 15.2, 7.6, 3.2 Hz, 1H), 1.74–1.59 (m, 2H), 1.15 (m, 1H), 0.98 (t, J = 7.2 Hz, 3H), 0.91 (s, 9H), 0.14 (s, 3H), 0.13 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ: 201.6, 167.7, 154.0, 108.5, 96.4, 51.4, 45.1, 43.6, 26.8, 25.8, 19.3, 18.1, 11.5, –4.2, –5.1.
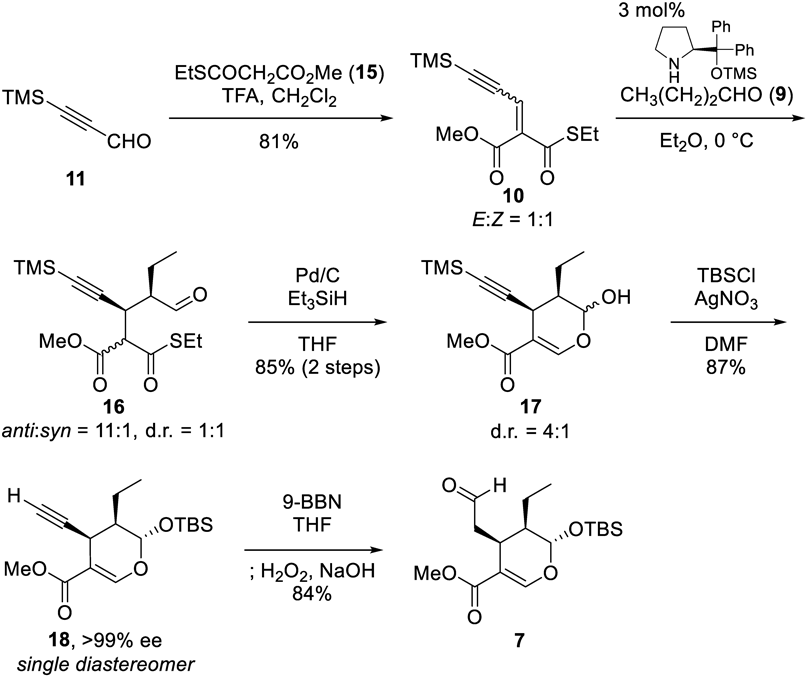
Preparation of Compound 19To a solution of secologanin derivative 7 (320 mg, 0.934 mmol), (R)-4-methoxy-α-cyanotryptamine (6, 201.1 mg, 0.934 mmol), and powdered MS 4 Å (1 g) in CH2Cl2 (18.7 mL) was added TFA (1.0 M in CH2Cl2, 467 µL, 0.467 mmol) at 0 °C under Ar atmosphere. The reaction mixture was stirred for 30 min at 0 °C. The resulting mixture was filtered through a cotton plug with CHCl3 and the filtrate was added to an excess amount of saturated aqueous NaHCO3 solution at 0 °C. The aqueous layer was extracted three times with CHCl3. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 15% AcOEt/n-hexane) afforded 19 (504.3 mg, quantitative yield) as a single diastereomer.
Compound 19: White amorphous powder; [α]23D –230.1 (c 0.85, CHCl3); IR (ATR) νmax cm−1 3328, 2928, 1688, 1630, 1572, 1509, 1463, 1437, 1384, 1357, 1312, 1279, 1254, 1207, 1167, 1137, 1102, 1029, 1018, 974, 938, 892, 871, 837; HRMS (ESI) [M + H]+ Calcd. for [C29H42N3O5Si1]+: 540.2894, Found: 540.2859; UV (MeOH) λmax 292, 225 nm; 1H-NMR (600 MHz, CDCl3) δ: 7.82 (br s, 1H), 7.59 (s, 1H), 7.04 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 7.8 Hz, 1H), 6.47 (d, J = 7.8 Hz, 1H), 5.26 (d, J = 9.0 Hz, 1H), 4.37 (dd, J = 4.2, 3.6 Hz, 1H), 4.07 (ddd, J = 10.8, 3.6, 1.8 Hz, 1H), 3.88 (s, 3H), 3.79 (s, 3H), 3.35–3.29 (m, 2H), 2.97 (dt, J = 12.0, 4.2 Hz, 1H), 1.79 (ddd, J = 12.6, 11.4, 3.0 Hz, 1H), 1.73–1.63 (m, 2H), 1.55 (td, J = 12.6, 2.4 Hz, 1H), 1.18 (m, 1H), 0.98 (t, J = 7.2 Hz, 3H), 0.93 (s, 9H), 0.19 (s, 3H), 0.18 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ: 169.3, 154.6, 154.4, 137.4, 132.4, 122.8, 120.0, 117.5, 109.0, 105.6, 104.3, 100.0, 97.2, 55.3, 52.2, 46.8, 44.4, 43.8, 36.9, 27.8, 26.9, 25.8, 20.2, 18.1, 11.1, –3.9, –4.9.
Preparation of 9-Methoxystrictosidine Derivative 5Acetic acid (265 µL, 4.63 mmol) and sodium cyanoborohydride (582 mg, 9.26 mmol) were added to a solution of 19 (500 mg, 0.936 mmol) in MeOH (4.6 mL) at r.t. under Ar atmosphere. The reaction mixture was stirred for 24 h at r.t. To the resulting mixture were added additional acetic acid (265 µL, 4.63 mmol) and sodium cyanoborohydride (582 mg, 9.26 mmol) under Ar atmosphere. The reaction mixture was stirred for a further 36 h at r.t. The resulting mixture was quenched with saturated aqueous NaHCO3 solution at 0 °C. The aqueous layer was extracted three times with AcOEt. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude materials were filtered through a short plug of amino-silica gel (SiO2-NH), eluted with 5% MeOH/CHCl3, to remove an excess amount of sodium cyanoborohydride. Flash chromatography (SiO2, 25% AcOEt/n-hexane then 20% MeOH/CHCl3) afforded the 9-methoxystrictosidine derivative 5 (371.6 mg, 78%) with 80.5 mg of starting material 19.
9-Methoxystrictosidine Derivative 5; White amorphous powder; [α]23D –189.9 (c 0.93, CHCl3); IR (ATR) νmax cm−1 2933, 2850, 1698, 1630, 1510, 1460, 1436, 1383, 1357, 1308, 1276, 1252, 1166, 1137, 1096, 1031, 1007, 975, 950, 927, 908, 872, 861, 836, 825; HRMS (ESI) [M + H]+ Calcd. for [C28H43N2O5Si1]+: 515.2941, Found: 515.2955; UV (MeOH) λmax 292, 223 nm; 1H-NMR (600 MHz, CDCl3) δ: 8.33 (br s, 1H), 7.55 (s, 1H), 7.02 (t, J = 7.8 Hz, 1H), 6.94 (d, J = 7.8 Hz, 1H), 6.46 (d, J = 7.8 Hz, 1H), 5.23 (d, J = 8.4 Hz, 1H), 4.01 (dd, J = 9.0, 3.0 Hz, 1H), 3.88 (s, 3H), 3.74 (s, 3H), 3.29 (m, 1H), 3.05–2.95 (m, 4H), 1.92 (ddd, J = 13.8, 10.8, 3.6 Hz, 1H), 1,71–1.61 (m, 2H), 1.52 (ddd, J = 13.8, 10.2, 3.6 Hz, 1H), 1.27 (m, 1H), 0.99 (t, J = 7.2 Hz, 3H), 0.92 (s, 9H), 0.17 (s, 3H), 0.16 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ: 169.3, 154.8, 154.6, 137.3, 133.6, 122.3, 117.5, 109.8, 108.6, 104.5, 99.9, 97.3, 55.4, 51.8, 50.5, 44.4, 42.1, 36.7, 28.0, 25.8, 24.2, 20.1, 18.1, 11.6, –4.0, –5.0.
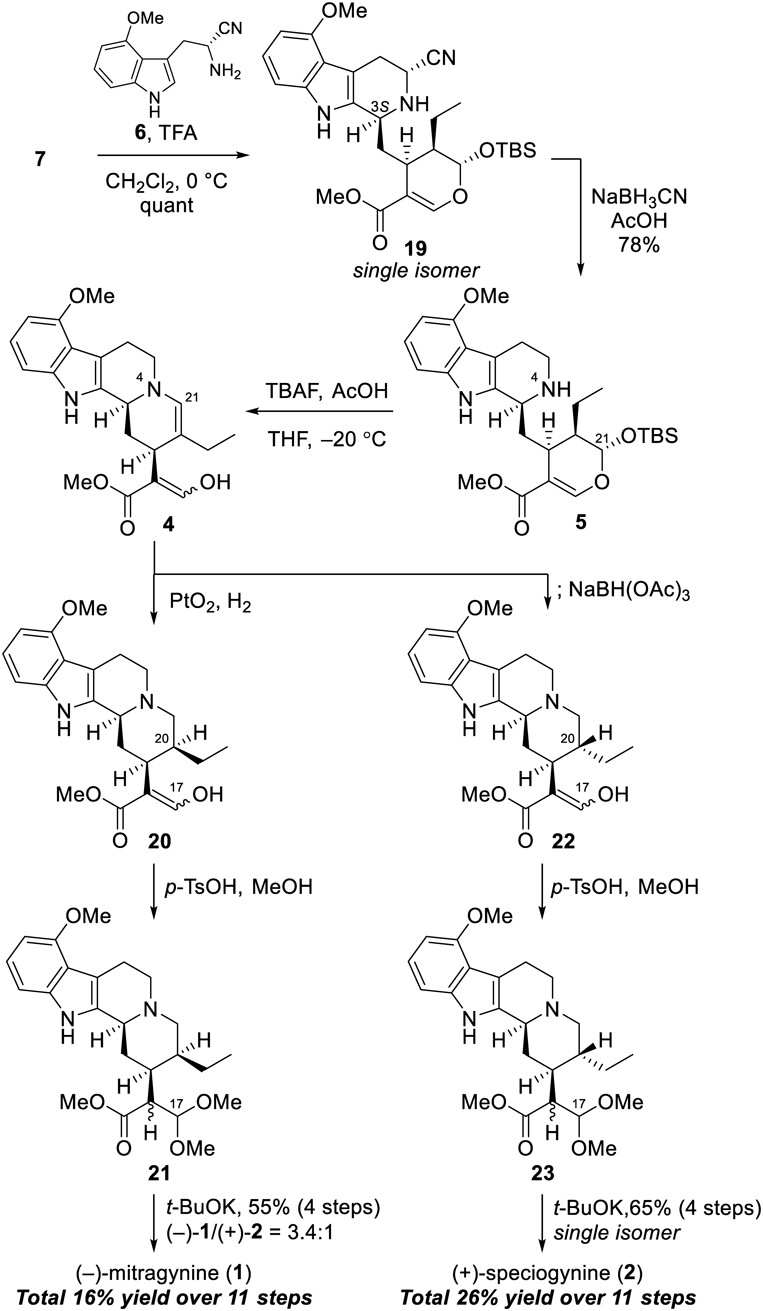
Synthesis of (−)-Mitragynine (1)To a solution of 9-methoxystrictosidine derivative 5 (150 mg, 0.291 mmol) in degassed anhydrous THF (2.9 mL) were added acetic acid (83.3 µL, 1.46 mmol) and tetrabutylammonium fluoride (1.0 M in THF solution, 583 µL, 0.583 mmol) at –20 °C under Ar atmosphere. The reaction mixture was stirred for 5 h at –20 °C. The resulting mixture was directly filtered through a short plug of silica gel eluted with 40% AcOEt/n-hexane. The residue of 4 was dissolved in degassed MeOH (14.6 mL) to which was added PtO2 (150 mg, 100 wt%) at r.t. under Ar atmosphere. The resulting mixture was purged with a stream of hydrogen and the reaction mixture was stirred for 1 h at r.t. under H2 atmosphere. The resulting mixture was filtered with a Celite pad with AcOEt, and the filtrate was concentrated under reduced pressure. The resulting residue of 20 was dissolved in degassed MeOH (14.6 mL) to which was added CH(OMe)3 (52.7 µL, 0.481 mmol) and p-TsOH·H2O (211 mg, 1.11 mmol), and the mixture was stirred at 70 °C for 16 h. The resulting mixture was quenched with saturated aqueous NaHCO3 solution, and was concentrated under reduced pressure. The aqueous layer was extracted three times with CHCl3. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. The crude materials of 21 were dissolved in dry degassed DMF (5.8 mL) to which was added t-BuOK (98.1 mg, 0.874 mmol) at 0 °C under Ar atmosphere. The reaction mixture was stirred for 45 min at r.t. under Ar atmosphere. The resulting mixture was quenched with water and the aqueous layer was extracted three times with CHCl3. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 25–45% AcOEt/n-hexane gradient then 5% MeOH/CHCl3) afforded (−)-mitragynine (1, 48.7 mg, 42% over four steps) and (+)-speciogynine (2, 15.2 mg, 13% over four steps). All spectral data of the obtained (−)-mitragynine (1) and (+)-speciogynine (2) were identical to those of natural products.33,34)
(−)-Mitragynine (1): Yellow amorphous powder; [α]24D –103.1 (c 0.57, CHCl3) [lit. [α]24D –126 (c 1.2, CHCl3)]; IR (ATR) νmax cm−1 3359, 2926, 1697, 1622, 1567, 1508, 1461, 1434, 1350, 1273, 1253, 1188, 1170, 1146, 1104, 1078, 1020, 977, 903, 885, 862; HRMS (ESI) [M + H]+ calculated for [C23H31N2O4]+: 399.2284, found: 399.2291; CD (0.3 mM, MeOH, 23 °C) λ nm (Δε): 320 (0), 291 (2.27), 283 (2.15), 273 (2.45), 261 (0), 239 (–8.18), 229 (–7.33), 219 (–11.32), 206 (0); UV (MeOH) λmax 292, 224 nm; 1H- and 13C-NMR see supporting information.
(+)-Speciogynine (2): Yellow amorphous powder; [α]24D +17.0 (c 1.02, CHCl3) [lit. [α]24D +26.8 (c 0.85, CHCl3)]; IR (ATR) νmax cm−1 3363, 2931, 2846, 2796, 2736, 1699, 1633, 1568, 1508, 1460, 1434, 1355, 1335, 1317, 1276, 1252, 1222, 1187, 1144, 1102, 1013, 989, 915, 894, 862, 834, 810; HRMS (ESI) [M + H]+ Calcd. for [C23H31N2O4]+: 399.2284, Found: 399.2273; CD (0.3 mM, MeOH, 23 °C) λ nm (Δε): 309 (0), 291 (2.93), 281 (3.56), 269 (4.47), 253 (3.36), 237 (9.55), 230 (0), 219 (–20.57), 203 (0); UV (MeOH) λmax 291, 225 nm; 1H- and 13C-NMR see supporting information.
Synthesis of (+)-Speciogynine (2)To a solution of 9-methoxystrictosidine derivative 5 (30 mg, 0.058 mmol) in degassed anhydrous THF (580 µL) were added acetic acid (16.7 µL, 0.291 mmol) and tetrabutylammonium fluoride (1.0 M in THF solution, 117 µL, 0.117 mmol) at –20 °C under Ar atmosphere. The reaction mixture was stirred for 5 h at –20 °C. To the resulting mixture was added sodium triacetoxyborohydride (24.7 mg, 0.117 mmol) at –20 °C. The reaction mixture was stirred for 2.5 h at –20 °C. The resulting mixture was quenched with saturated aqueous NaHCO3 solution and the aqueous layer was extracted three times with AcOEt. The combined organic layer was washed with brine then dried over Na2SO4. The resulting residue of 22 was dissolved in degassed MeOH (2.9 mL), to which was added CH(OMe)3 (10.5 µL, 0.0962 mmol) and p-TsOH·H2O (42.1 mg, 0.221 mmol), and then stirred at 70 °C for 12 h. The resulting mixture was quenched with saturated aqueous NaHCO3 solution and then concentrated under reduced pressure. The aqueous layer was extracted three times with CHCl3. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. The crude materials of 23 were dissolved in dry degassed DMF (1.2 mL) to which was added t-BuOK (19.6 mg, 0.175 mmol) at 0 °C under Ar atmosphere. The reaction mixture was stirred for 40 min at r.t. under Ar atmosphere. The resulting mixture was quenched with water and the aqueous layer was extracted five times with CHCl3. The combined organic layer was dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2, 45% AcOEt/n-hexane) afforded (+)-speciogynine (2, 15.2 mg, 65% over four steps).
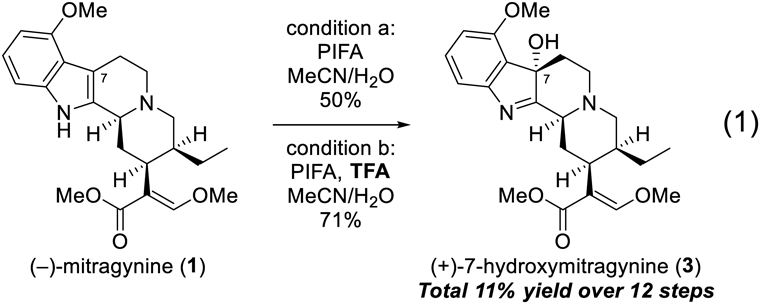
Synthesis of (+)-7-Hydroxymitragynine (3)To a solution of freshly purified (−)-mitragynine (1, 30 mg, 0.075 mmol) in MeCN (900 µL) and water (100 µL) was added TFA (11.5 µL, 0.151 mmol) at 0 °C, and the reaction mixture was stirred for 5 min at 0 °C. PIFA (32.4 mg, 0.0753 mmol) was added to the stirred mixture at 0 °C under Ar atmosphere. The reaction mixture was stirred for 20 min at 0 °C, followed by quenching with saturated aqueous NaHCO3 solution. The aqueous layer was extracted three times with CH2Cl2. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. Flash chromatography (SiO2-NH, 40% AcOEt/n-hexane) afforded (+)-7-hydroxymitragynine (3, 22.0 mg, 71%). All spectral data of the obtained (+)-7-hydroxymitragynine (3) were identical to those of natural product.6,8)
(+)-7-Hydroxymitragynine (3): Pale yellow amorphous powder; [α]23D +62.5 (c 0.50, CHCl3) [lit. [α]23D +47.9 (c 0.55, CHCl3)]; IR (ATR) νmax cm−1 3420, 2947, 2872, 2835, 2811, 2745, 1698, 1644, 1597, 1486, 1460, 1435, 1376, 1266, 1237, 1187, 1143, 1104, 1074, 1018, 989, 959, 935, 920, 870, 841; HRMS (ESI) [M + H]+ Calcd. for [C23H31N2O5]+: 415.2233, Found: 415.2256; CD (0.3 mM, MeOH, 23 °C) λ nm (Δε): 344 (0), 305 (0.53), 282 (0), 258 (0.82), 243 (0), 229 (−1.13), 214 (0); UV (MeOH) λmax 307, 245, 220 nm; 1H- and 13C-NMR see supplementary materials.