Results and Discussion
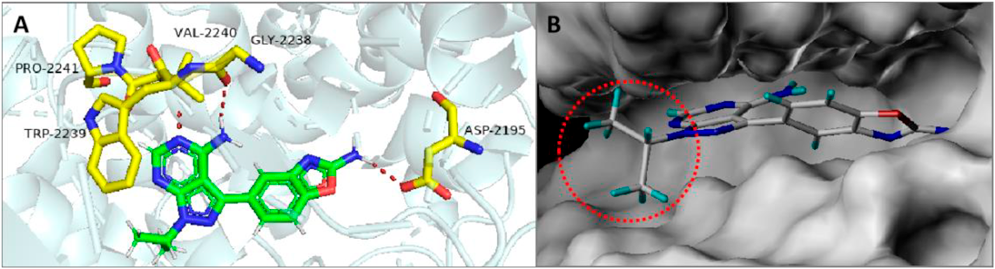
Design StrategyThe compound MLN0128 was molecularly docked with the mTOR protein (PDB ID: 4JT5) (Fig. 2A). It was observed that a hydrogen bond was formed between the pyrimidine ring and Val2240 and Gly2238 of the hinge region. The benzoxazole ring moiety formed hydrogen bonds with the Asp2195 residue in the hydrophobic pocket, while the N-1 position of the pyrazole ring of MLN0128 had no obvious interaction with any amino acid. Therefore, we speculated that the introduction of a linker at this position had little effect on its binding to mTOR. In addition, the docking results showed that the N-1 position of the pyrazole ring of MLN0128 was exposed to the solvent (Fig. 2B), suggesting that this position can serve as a suitable attachment site for the linker. In this study, we designed and synthesized three mTOR-targeting PROTACs (P1–P3, Fig. 3) by introducing the E3 ligase ligand pomalidomide into the N-1 position of the pyrazole ring of MLN0128 through different linkers.
ChemistryThe synthesis of the target compound is shown in Chart 1. Using the halogenated compound of pyrazolo[3,4-d]pyrimidin-4-amine as the raw material, the pyrazole ring N-1-substituted MLN0128 derivatives 5, 8, and 11 were obtained by Suzuki coupling and substitution reactions. Compound 8 was deprotected to generate compound 9. Compound 11 was reduced to generate compound 12. According to the method previously reported, using pomalidomide 13 as the raw material, the azide derivative 15 and the amino derivative 16 of pomalidomide were obtained by acylation, azide, and reduction reactions. A substitution reaction between pomalidomide and Br-PEG3-t-butyl ester 17 generated compound 18. The click reaction of intermediate 5 with azide 15 afforded compound P1. The condensation of intermediates 12 and 16 afforded compound P2. The condensation of intermediates 9 and 18 afforded compound P3. The structures of the target compounds were characterized by 1H-NMR, 13C-NMR, and high resolution (HR)MS.
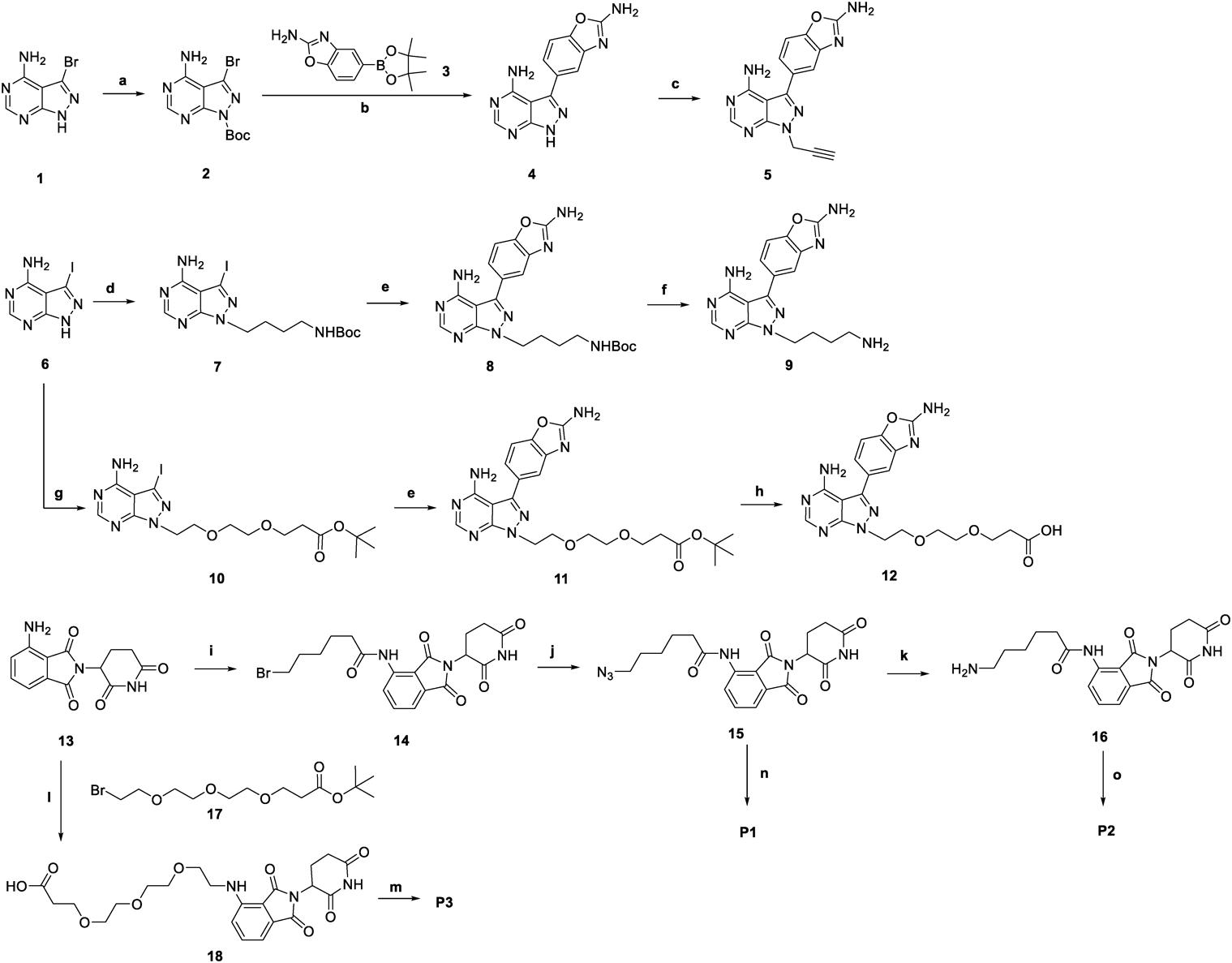
Chart 1. Syntheses of Compounds
P1–
P3Reagents and conditions: (a) Boc2O, N,N-dimethyl-4-aminopyridine (DMAP), dichloromethane (DCM), room temperature (r.t.), 12 h, 56%; (b) Pd(PPh3)4, Na2CO3, dioxane/H2O, 90 °C, 12 h, 57%; (c) 3-bromoprop-1-yne, Na2CO3, N,N-dimethylformamide (DMF), r.t., 12 h, 68%; (d) tert-butyl (4-bromobutyl)carbamate, K2CO3, DMA, r.t., 3 h, 46%; (e) 3, Pd(PPh3)4, Na2CO3, l,2-dimethoxyethane (DME)/H2O = 2 : 1, 110 °C, 12 h, 35-49%; (f) gas HCl/MeOH (4 N), r.t., 4 h, 99%; (g) tert-butyl 3-(2-(2-bromoethoxy)ethoxy)propanoate, K2CO3, DMA, 80 °C, 3 h, 62%; (h) trifluoroacetic acid (TFA), Et3SiH, DCM, r.t., 12 h, 70%; (i) 6-bromohexanoyl chloride, tetrahydrofuran (THF), 75 °C, 2 h, 78%; (j) NaN3, DMF, 90 °C, 12 h, 99%; (k) PPh3, THF/1 M HCl aq, r.t., 4 h, 52%; (l) 17, K2CO3, DMF, 75 °C, 24 h; TFA, CH2Cl2, r.t., 2 h, 46%; (m) 9, HATU, triethylamine (TEA), DMF, r.t., 12 h, 39%; (n) 5, CuSO4·5H2O, Sodium ascorbate, THF/H2O (10 : 1), r.t., 12 h, 28%; (o) 12, DIEA, HATU, DMF, r.t., 4 h, 43%.
To assess the binding of synthetic compounds to mTOR, kinase activity assays were performed. The inhibitory rates of compounds P1–P3 on mTOR are presented in Table 1. Compounds P1–P3 had inhibitory activity on mTOR, which confirmed our hypothesis, indicating that the N-1 position of the pyrazole ring of MLN0128 is a suitable attachment site for the introduction of a linker which has little effect on the binding capacity of mTOR. Compound P1 had the best inhibitory activity on mTOR, with IC50 of 0.087 µM, and the activities of compounds P2 and P3 with extended linkers were decreased to varying degrees, indicating that the linker of PROTACs should not be too long.
Table 1. Inhibition Activities of Compounds against mTOR
| Compounds | IC50 (µM) |
|---|
| P1 | 0.087 ± 0.004 |
| P2 | 0.13 ± 0.013 |
| P3 | 0.35 ± 0.065 |
| MLN0128 | 0.005 ± 0.001 |
To elucidate the effect of triazole ring on the activity, molecular docking studies were carried out by using Surflex-Dock module of SYBYL-X 2.0 (Tripos). Compound P1 was docked to the crystal structure of mTOR (PDB ID code: 4JT5). The docking result (Fig. 4) indicated that the “warhead” of P1 formed two hydrogen bonds and hydrophobic interactions with ATP-binding site of mTOR, and there was no apparent interaction between the triazole ring with mTOR.
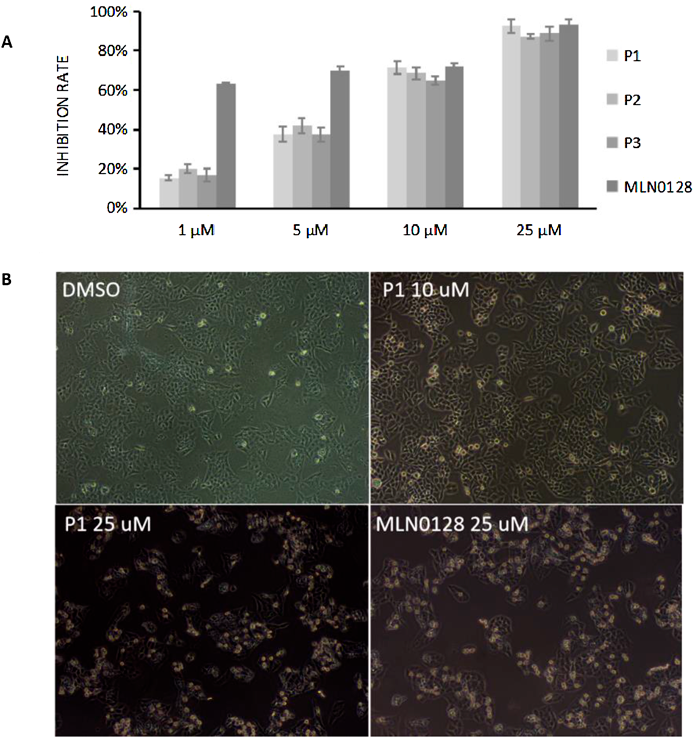
Anti-proliferative ActivityThe anti-proliferative effects of the target compounds P1–P3 on human breast cancer MCF-7 cells was evaluated by MTT colorimetry. The results (Fig. 5A) showed that the anti-proliferative effects of P1–P3 were all concentration-dependent, and the inhibition rate at 25 µM (72 h of treatment) reached 90%. Compound P1 exhibited comparable anti-proliferative activity against MCF-7 cells with MLN0128 at 10 and 25 µM. P1 has an IC50 of 0.087 µM in the biochemical assay (Table 1) but the potency was not translated into cell growth inhibition (roughly 100-fold shift). This is probably because the compound is too polar to penetrate the cell membrane (polar bonds exist in both ends). After observing the effects of different concentrations of P1 on the state of MCF-7 cells, it was observed that, compared with the dimethyl sulfoxide (DMSO) controls, the changes in the state of the cells were not obvious after treatment with 10 µM P1 for 6 h, while the cells shrank significantly and the cell proliferation rate was decreased after treatment with 25 µM P1 for 6 h (Fig. 5B). It might be due to autophagy-mediated cell death and a similar phenomenon was in accordance with reported PROTACs for degradation of PI3K.22) Compound P1, which showed excellent mTOR kinase inhibitory activity and anti-proliferative activity, was selected for further evaluation.
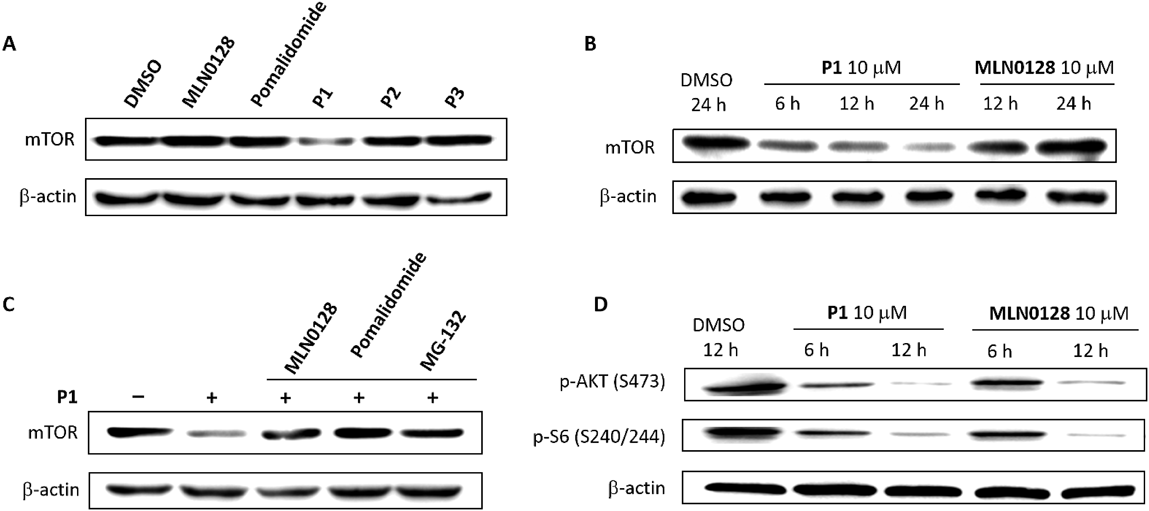
Degradation of mTOR and Influence on mTOR Downstream ProteinTo verify the ability of P1 to degrade mTOR, we first examined the effects of MLN0128 and pomalidomide on mTOR levels in MCF-7 cells and found that neither of these treatments affected the levels of mTOR (Fig. 6A). It was confirmed that MLN0128 or pomalidomide could not induce mTOR degradation in MCF-7 cells.
The degradation levels of mTOR in MCF-7 cells treated with P1, P2 and P3 were detected preliminarily by Western blotting (Fig. 6A). Compared with control DMSO-treated cells, MCF-7 cells treated with P1 showed an obvious decrease of the mTOR level, but unfortunately cells treated with P2 or P3 showed an insignificant decrease of mTOR, which suggested that P2 and P3 exhibited lower capacity of mTOR degradation than P1. The results once again confirmed the conclusion that the linker of PROTACs should not be too long.
Cells treated with P1 for different times were analyzed by Western blotting (Fig. 6B). It was demonstrated that compound P1 could degrade mTOR in a time-dependent manner. Compared with controls which were treated with DMSO, MCF-7 cells treated with P1 for 6 h showed an obvious decrease of the mTOR level, and the degradation was more significant after treatment for 24 h. The degradation of mTOR was not observed obviously after treatment with MLN0128, possibly because MLN0128 could inhibit mTOR activity but not degrade mTOR protein.
To determine whether P1 induces degradation of mTOR via the ubiquitin-proteasome system, MCF-7 cells were treated with the proteasome inhibitors MG-132 prior to P1 application (Fig. 6C). It was found that inhibition of proteasome effectively blocked the P1-induced degradation of mTOR, indicating that this degradation depends on the ubiquitin-proteasome system.
The changes in the expression levels of the mTOR downstream proteins p-S6 (Ser240/244) and p-AKT (Ser473) in MCF-7 cells treated with compound P1 were examined by Western blotting. As shown in Fig. 6D, compared with controls which were treated with DMSO, the expression levels of p-AKT (Ser473) and p-S6 (Ser240/244) decreased with the prolongation of the P1 treatment time, which was similar to the results of MLN0128, indicating that compound P1 can inhibit the mTOR downstream pathway in cells.
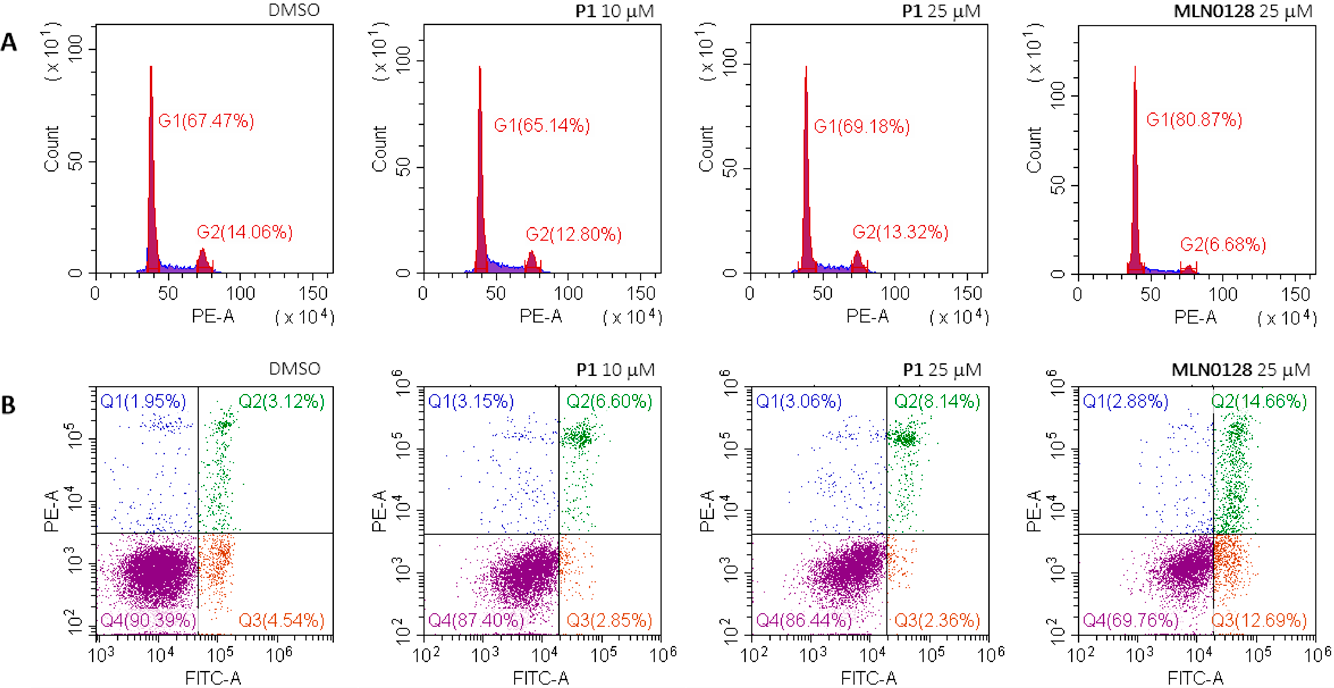
Effects on Cell Cycle and ApoptosisTo further explore the mechanism of action of these compounds, the effects of compound P1 on the cell cycle and apoptosis of MCF-7 cells was examined by flow cytometry. The results (Fig. 7) showed that after P1 treatment for 24 h, compared with blank controls, the cell cycle and apoptosis of MCF-7 cells did not change significantly. These effects were different from those of MLN0128, which induced G1 cell cycle arrest and apoptosis.
Effects on AutophagyConsidering that mTOR is related to autophagy, we examined the conversion of the autophagy marker proteins LC3A/B-I and LC3A/B-II by Western blotting. As shown in Fig. 8, the expression level of LC3A/B-II in MCF-7 cells increased with the prolongation of the P1 treatment time compared to the DMSO controls. And the promoted LC3A/B-II expression levels by P1 were better than positive control MLN0128. The results indicated that P1 could induce autophagy in MCF-7 cells. Although both MLN0128 and P1 inhibit mTOR and its downstream proteins, the mechanisms of action are different. MLN0128 hinders the binding of ATP to mTOR, thereby inhibiting the activation of mTOR. P1 degrades mTOR, which reduces mTOR, and at the same time, may trigger autophagy during the degradation process. More evidence is still needed for the autophagy-inducing effect of compound P1.
Physiochemical Property PredictionsTo explore the potential of P1 for bioavailability, we evaluated several physicochemical descriptors of P1 associated with compound permeability/absorption including molecular weights (MW), lipophilicities (c Log P), H-bond donors (HBD), H-bond acceptors (HBA), the number of rotatable bonds (nRotB) and topological polar surface areas (TPSA) (Table 2). PROTACs are heterobifunctional compounds with molecular weights and other properties that lie in chemical space beyond that described by Lipinski’s rule-of-five (Ro5) and/or Veber’s rotatable bond/polar surface area guidelines.32) Then the drug likeliness of compound P1 was evaluated on the basis of Kihlberg’s beyond Ro5 (bRo5) and AbbVie’s bRo5,33,34) in which the bRo5 compounds often possess molecular weights above 700 and/or other violations of Lipinski/Veber’s rules.
Table 2. Physiochemical Properties
a) Table for Compound
P1| Compound | Mw | c Log P | HBD | HBA | nRotB | TPSA (Å2) |
|---|
| P1 | 717.69 | 1.3 | 4 | 12 | 12 | 265.03 |
a) Physiochemical property predictions of P1 were calculated using SwissADME (swissadme.ch). The c Log P values represent the average of five Log P predictions from the programme.
The values for in silico descriptors of P1 were summarized on a radar plot where we compared the P1 properties to the upper boundary limits for sets of Ro5 and bRo5 compounds (Fig. 9). The results showed that the physicochemical properties of P1 did not fall within the Ro5 space but falled within bRo5 space. P1 was close to drug-like space with the MW, c Log P, HBD, HBA and nRotB values falling within the bRo5 boundaries, but still suffered from TPSA higher than the upper boundary. Further optimization of compound P1 to reduce TPSA may help move it into even more promising space.
Experimental
MaterialsCommon chemicals and solvents used were obtained from commercial sources. The solvents were dried by standard procedures. Thin layer chromatography (Silica gel GF254) and silica column chromatography (Silica gel 200–300 mesh) were obtained from Qingdao Haiyang Chemical Co., Ltd., Qingdao, China. 1H-NMR spectra and 13C-NMR spectra were recorded at 400 or 600 MHz using Bruker Avance III spectrometer in CDCl3 or DMSO-d6 solution. The NMR data was processed by software MestReNova. The high-resolution mass spectra (HRMS) was measured on Agilent 1260HPLC-1290 UPLC/G6230B TOF mass spectrometer. The mass spectra (electrospray ionization (ESI)-MS) was measured on Agilent 1260HPLC/6420 mass spectrometer.
Chemistry and Chemical MethodsCompounds 7, 8, 9 and pomalidomide derivatives 14, 15, 16 were synthesized according to the literatures.5,22)
5-(4-Amino-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzo[d]-oxazol-2-amine (4)To the solution of 3-bromo-1H-pyrazolo[3,4-d]pyrimidin-4-amine 1 (5.0 g, 23.3 mmol) and DMAP (0.28 g, 0.01 mmol) in DCM (100 mL), Boc2O (5.58 g, 25.6 mmol) was added. The reaction mixture was stirred at room temperature for 12 h and then concentrated to remove DCM. Then the residue was washed with a mixture of methyl tert butyl ether and ethyl acetate (MTBE/EtOAc = 5 : 1) to afford 2 (4.1 g, 56% yield) as an off-white solid. The solution of compound 2 (1 g, 3.18 mmol), 2-aminobenzo[d]oxazol-5-ylboronic acid pinacol ester (1.25 g, 4.78 mmol), Na2CO3 (0.67 g, 6.36 mmol) and Pd(PPh3)4 (0.3 g, 0.26 mmol) in a mixture of dioxane and water (50 mL, 10 : 1) was heated at 90 °C for 12 h under nitrogen. After cooling to room temperature, the reaction mixture was washed with water, extracted with ethyl acetate. The organic layer was dried (Na2SO4) and evaporated under vacuum to give crude product 4 (0.48 g, 57% yield) a gray solid. 1H-NMR (400 MHz, DMSO-d6) δ: 8.18 (s, 1H), 7.57 (s, 2H), 7.46 (d, J = 8.1 Hz, 1H), 7.41 (s, 1H), 7.23 (d, J = 8.1 Hz, 1H). The crude product was used in the next step without any further purification.
5-(4-Amino-1-(prop-2-yn-1-yl)-1H-pyrazolo[3,4-d]-pyrimidin-3-yl)benzo[d]oxazol-2-amine (5)To a solution of compound 4 (0.45 g, 1.68 mmol) and Na2CO3 (0.35 g, 3.37 mmol) in DMF (10 mL) was added 3-bromoprop-1-yne (0.80 g, 6.74 mmol), the resulting solution stirred for 12 h at room temperature. After the reaction completed, the reaction mixture was washed with water, extracted with ethyl acetate. The organic layer was dried (Na2SO4) and evaporated under vacuum to give crude product 5 (0.35 g, 68% yield). 1H-NMR (600 MHz, DMSO-d6) δ: 8.28 (s, 1H), 7.57 (s, 2H), 7.48 (d, J = 8.0 Hz, 1H), 7.41 (s, 1H), 7.25 (d, J = 8.1 Hz, 1H), 5.19 (s, 2H), 3.39 (s, 1H). The crude product was used in the next step without any further purification.
tert-Butyl-(2-(2-(4-amino-3-iodo-1H-pyrazolo[3,4-d]-pyrimidin-1-yl)ethoxy)ethoxy)propanoate (10)To a solution of compound 6 (0.26 g, 1 mmol) and tert-butyl-3-(2-(2-bromoethoxy)ethoxy)propanoate (0.25 g, 0.85 mmol) in DMA (4 mL) was added K2CO3 (0.36 g, 2 mmol). The mixture was heated at 80 °C for 3 h. After cooling to room temperature, the mixture was washed with water, extracted with MTBE. The combined organic layer was concentrated and the residue was recrystallized from MTBE to give 10 (0.25 g, 62% yield) as a yellow solid. m.p. 127-128 °C; 1H-NMR (600 MHz, DMSO-d6) δ: 8.19 (s, 1H), 4.41 (t, J = 5.5 Hz, 2H), 3.81 (t, J = 5.6 Hz, 2H), 3.51–3.43 (m, 4H), 3.40–3.36 (m, 2H), 3.34 (s, 2H), 2.34 (t, J = 6.1 Hz, 2H), 1.36 (s, 9H).
tert-Butyl-3-(2-(2-(4-amino-3-(2-aminobenzo[d]oxazol-5-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethoxy)ethoxy)-propanoate (11)Similar procedure of 8 was performed to give compound 11 (0.1 g, 35% yield) as a white solid. m.p. 189–190 °C; 1H-NMR (600 MHz, DMSO-d6) δ: 8.24 (s, 1H), 7.54 (s, 2H), 7.47 (d, J = 8.1 Hz, 1H), 7.41 (d, J = 1.5 Hz, 1H), 7.24 (dd, J = 8.1, 1.7 Hz, 1H), 4.47 (t, J = 5.7 Hz, 2H), 3.88 (t, J = 5.7 Hz, 2H), 3.53–3.46 (m, 4H), 3.43–3.40 (m, 2H), 2.32 (t, J = 6.2 Hz, 2H), 1.35 (s, 9H). ESI-MS m/z C23H29N7O5 [M + H]+: 484.10, [M + Na]+: 506.10.
3-(2-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)propanoic Acid (18)To a solution of 13 (0.15 g, 0.55 mmol) and Bromo-PEG4-t-butyl ester 17 (0.22 g, 0.65 mmol) in dry DMF (3 mL), K2CO3 (0.09 g, 0.65 mmol) was added. The mixture was heated at 75 °C for 24 h. After the reaction was completed, the mixture was cooled to room temperature and washed with water, extracted with EtOAc. The organic layer was dried (Na2SO4) and evaporated under vacuum. To the solution of the residue in DCM (5 mL) was added TFA (3 mL), the reaction mixture was stirred at room temperature for 2 h and then concentrated. Then the residue was redissolved in water and washed with EtOAc and MTBE. The aqueous phase was dried by vacuum freeze-drying to afford 18 (0.12 g, 46% yield). The characterization data were identical with those reported in literature.35)
6-(4-((4-Amino-3-(2-aminobenzo[d]oxazol-5-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-hexanamide (P1)To a mixture of compound 5 (90 mg, 0.30 mmol), 15 (121 mg, 0.30 mmol) and CuSO4 ∙ 5H2O (49 mg, 0.20 mmol) in a mixture of THF and water (5 mL, 10 : 1) was added sodium ascorbate (5.8 mg, 0.03 mmol).
The resulting solution stirred for 12 h at room temperature. After the reaction completed, the reaction mixture was washed with water, extracted with ethyl acetate. The organic layer was dried (Na2SO4) and evaporated under vacuum. The residue was purified by preparative HPLC (cloumn: Phenomenex Luna 80*30 mm*3um; mobile phase: (water-ACN); B%: 15%–45%, 8 min) to afford the desired compound P1 (60 mg, 28% yield) as a white solid. m.p. 200–202 °C; 1H-NMR (600 MHz, DMSO-d6) δ: 11.15 (s, 1H), 9.70 (s, 1H), 9.29 (s, 1H), 8.62 (s, 1H), 8.51 (s, 1H), 8.44 (d, J = 8.4 Hz, 1H), 8.19 (s, 1H), 7.85–7.80 (m, 1H), 7.61 (t, J = 8.0 Hz, 2H), 7.50 (s, 1H), 7.34 (d, J = 8.2 Hz, 1H), 5.70 (s, 2H), 5.14 (dd, J = 12.9, 5.4 Hz, 2H), 4.33 (t, J = 7.1 Hz, 2H), 2.95–2.84 (m, 1H), 2.61 (dd, J = 14.2, 2.8 Hz, 1H), 2.57–2.51 (m, 1H), 2.44 (t, J = 7.4 Hz, 2H), 2.10–2.02 (m, 1H), 1.88–1.79 (m, 2H), 1.67–1.59 (m, 2H), 1.33–1.25 (m, 2H). 13C-NMR (151 MHz, DMSO-d6) δ: 172.79, 171.86, 169.81, 167.62, 166.68, 152.72, 151.57, 147.87, 146.85, 141.46, 136.47, 136.10, 131.48, 127.21, 126.42, 124.01, 121.85, 118.37, 117.11, 114.28, 109.96, 96.80, 49.28, 48.90, 42.50, 36.17, 30.94, 29.41, 25.35, 24.10, 21.99, 0.15, 0.12. HRMS Calcd for C34H32N13O6 [M + H]+ m/z 718.2659, Found 718.2662.
6-(3-(2-(2-(4-Amino-3-(2-aminobenzo[d]oxazol-5-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethoxy)ethoxy)propanamido)-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)-hexanamide (P2)To a solution of 11 (0.2 g, 0.41 mmol) in TFA (2 mL), catalytic amount of triethylsilane were added. The mixture was then stirred at room temperature for 12 h and then concentrated. The residue was washed with a mixture of methyl tert butyl ether and ethyl acetate (MTBE/PE = 1 : 2) to afford 12 (0.12 g, 70% yield). The crude product was used in the next step without any further purification. To a mixture of compound 12 (60 mg, 0.14 mmol), 16 (54 mg, 0.14 mmol) and HATU (79.8 mg, 0.21 mmol) in DMF (3 mL) was added TEA (29 mg, 0.28 mmol). The resulting solution stirred for 12 h at room temperature. After the reaction completed, the reaction mixture was purified by preparative HPLC (cloumn: Phenomenex Luna 80*30 mm*3 μm; mobile phase: (water(HCl)-ACN); B%: 15–45%, 8 min) to afford the desired compound P2 (48 mg, 43% yield) as a white solid. m.p. 153–154 °C; 1H-NMR (600 MHz, DMSO-d6) δ: 11.15 (s, 1H), 9.69 (s, 1H), 8.70 (s, 2H), 8.59 (s, 1H), 8.45 (d, J = 8.3 Hz, 1H), 7.86–7.78 (m, 2H), 7.62 (dd, J = 14.0, 7.6 Hz, 2H), 7.53 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 5.17–5.12 (m, 1H), 4.57 (s, 2H), 3.91 (t, J = 4.7 Hz, 2H), 3.53–3.46 (m, 4H), 3.39 (s, 2H), 3.04–2.98 (m, 2H), 2.95–2.82 (m, 4H), 2.61 (d, J = 16.2 Hz, 1H), 2.45 (t, J = 7.3 Hz, 2H), 2.22 (t, J = 6.2 Hz, 2H), 2.10–2.04 (m, 1H), 1.64–1.56 (m, 2H), 1.42–1.36 (m, 2H), 1.34–1.27 (m, 2H).13C-NMR (151 MHz, DMSO-d6) δ: 172.80, 171.97, 169.82, 169.77, 167.69, 166.68, 161.94, 160.86, 152.46, 151.94, 147.61, 147.23, 146.61, 136.53, 136.12, 131.46, 127.40, 126.30, 122.08, 118.32, 117.01, 114.09, 110.15, 96.60, 69.42, 69.36, 67.88, 66.85, 48.92, 46.94, 38.28, 36.47, 36.12, 30.95, 28.86, 25.93, 24.50, 22.00. HRMS Calcd for C38H42N11O9 [M + H]+ m/z 796.3231, Found 796.3235.
N-(4-(4-Amino-3-(2-aminobenzo[d]oxazol-5-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)butyl)-3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)-ethoxy)ethoxy)propanamide (P3)Using intermediate 9 (0.1 g, 0.29 mmol) and 18 (138 mg, 0.29 mmol), following the procedure to prepare P2, P3 was prepared. The reaction mixture was purified by preparative HPLC (cloumn: Phenomenex Luna 80*30 mm*3 μm; mobile phase: (water(HCl)-ACN); B%: 10–40%, 8 min) to afford the desired compound P3 (90 mg, 39% yield) as a white solid. m.p. 130–131 °C; 1H-NMR (600 MHz, DMSO-d6) δ: 11.09 (s, 1H), 9.77–9.43 (m, 4H), 8.62 (s, 1H), 8.20 (s, 1H), 7.93 (t, J = 5.4 Hz, 1H), 7.71 (d, J = 8.3 Hz, 1H), 7.59–7.54 (m, 2H), 7.45 (dd, J = 8.3, 1.4 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 7.0 Hz, 1H), 5.06–5.02 (m, 1H), 4.41 (t, J = 6.4 Hz, 2H), 3.60 (t, J = 5.4 Hz, 2H), 3.55–3.52 (m, 4H), 3.50–3.47 (m, 2H), 3.46–3.43 (m, 4H), 3.43–3.40 (m, 2H), 3.07–3.02 (m, 2H), 2.91–2.83 (m, 1H), 2.61–2.54 (m, 2H), 2.26 (t, J = 6.4 Hz, 2H), 2.04–1.99 (m, 1H), 1.88–1.82 (m, 2H), 1.41–1.34 (m, 2H). 13C-NMR (151 MHz, DMSO-d6) δ: 172.94, 170.18, 170.15, 169.00, 167.38, 160.74, 152.36, 151.37, 146.81, 146.67, 146.46, 146.26, 136.34, 135.40, 132.12, 128.02, 123.30, 117.55, 113.42, 110.92, 110.77, 109.26, 96.67, 69.83, 69.80, 69.58, 68.94, 66.91, 48.63, 46.95, 41.77, 37.80, 36.21, 31.06, 26.43, 26.20, 22.23. HRMS Calcd for C38H44N11O9 [M + H]+ m/z 798.3363, Found 798.3370.
BioassayCell growth inhibition assay: The MCF-7 cancer cells were purchased from Procell Life Science & Technology Co. Ltd. (Wuhan, China). MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) medium (Boster, PYG0072) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin and 100 mg/mL streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. Cells were seeded in 96-well plates at a density of 4 × 103 per well overnight. Cells were exposed to the indicated compounds at different concentrations for 72 h.
Twenty microliters 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagents (5 mg/mL, Sangon Biotech, CB0331) was added to each well and the plates were incubated at 37 °C for another 4 h. Then DMEM was removed and 100 µL DMSO solution was add to each well to dissolve formazan crystals for 5 min. The optical density (OD) values were read at 490 nm. The experiment was repeated at least twice independently with duplicate measurements.
Enzymatic inhibition assays: The compounds were diluted in 10% DMSO and 5 µL of the dilution was added to a 50 µL reaction buffer. The reaction buffer contains 40 mM Tris, pH 7.4. 10 mM MgCl2, 0.1 mg/mL bovine serum albumin (BSA), 1 mM dithiothreitol (DTT), 10 µM ATP, mTOR kinase and the enzyme substrate. The enzymatic reactions were conducted at 30 °C for 40 min. It measures kinase activity by quantitating the amount of ATP remaining in solution following a kinase reaction by Kinase-Glo Plus luminescence kinase assay kit (Promega, V3771). Values were obtained by nonlinear regression analysis with GraphPad Prism.
Western blot analysis: MCF-7 cells were treated with P1 in different conditions at concentration of 10 µM. After compound treatment, cells were collected and washed before lysed by radio immunoprecipitation assay (RIPA) lysate buffer with protease and phosphatase inhibitor (Boster, AR1192, AR0102). Cell extracts were normalized for protein content with BCA protein assay kit (Boster, 15E28C46). Homogenates were centrifuged for 30 min at 4 °C, and the supernatants were collected and boiled in sodium dodecyl sulfate (SDS) loading buffer. Total protein was separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE), transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, IPVH00010, ISEQ00010). The membranes were blocked with 5% (w/v) milk in TBST buffer for 1 h, then incubated with primary antibody dilution overnight at 4 °C. After washing in TBST buffer, membrane was incubated in secondary antibodies for 0.5 h at room temperature. The membrane was washed again in TBST before they were imaged.
Flow cytometric analysis: MCF-7 cells were treated with P1 in different conditions. After compound treatment, cells were collected and washed. Apoptosis was determined using an Annexin V-FITC/PI Apoptosis Detection Kit (Boster, MK1028) and cell cycle distribution was determined by Cell Cycle and Apoptosis Analysis Kit (Yuanye, R20288).
Docking StudiesMolecular docking studies were performed using Surflex-Dock module of SYBYL-X 2.0 (Tripos). The crystal structure of mTOR complex (PDB code: 4JT5) was obtained from the RCSB Protein Data Bank. With the water molecules deleted, hydrogen added, ligand removed and force field applied, the receptor was used for the subsequent docking studies. The active site was identified by picking out the original co-crystalized ligand. For the preparation of ligand, 3D structures were generated and energy minimized by using Minimize module. The prepared ligands were docked into the active site of mTOR receptor using Surflex-Dock module. Images depicting the proposed binding modes were generated using PyMOL or SYBYL.