Regular Articles
Discovery of Indole-Piperazine Hybrid Structures as Potent Selective Class I Histone Deacetylases Inhibitors
2023 年 71 巻 3 号 p. 206-212
詳細
2023 年 71 巻 3 号 p. 206-212
Histone deacetylases (HDACs) are important targets in cancer treatment, and the development of selective and broad-spectrum HDACs inhibitors (HDACis) is urgent. In this research, a series of aroylpiperazine hybrid derivatives were designed and synthesized. Among these, indole-piperazine hybrids 6a (IC50 = 205 nM) and 6b (IC50 = 280 nM) showed submicromolar activity against HDAC1. Moreover, 6a showed a preferable affinity toward class I HDACs, especially for HDAC1–3. In vitro, 6a exhibited better antiproliferative activities against K562 and HCT116 cell lines than chidamide.
Histone deacetylases (HDACs) remove acetyl groups from lysine residues located in the amino radical terminal tails of core histones, and play an important role in both transcriptional and post-translational modifications.1,2) The eighteen HDAC isozymes contain class I (HDACs 1, 2, 3, and 8), class IIa (HDACs 4, 5, 7, and 9), class IIb (HDACs 6 and 10), and class IV (HDAC 11).3) Class I HDACs, especially for HDAC1, is highly expressed in various cancer cell lines and promotes malignant growth.4,5) By so far, a lot of synthetic HDACis have entered clinical trials, and several of them obtained approvals for cancer treatment such as vorinostat,6) belinostat,7) panobinostat,8) and chidamide.9,10) Generally, the pharmacophores of HDACs inhibitors (HDACis) include an aromatous cap, linker, and a zinc binding group (ZBG).11–15) Hydroxamic acid and benzamide are common ZBGs. Despite of higher potency, nonselective hydroxamic acid HDACis usually led to severe side effects,16) while selective benzamide HDACis could avoid this risk.4,17) The first clinical benzamide HDACi, MS-275 (Fig. 1), could selectively inhibited class I HDACs, with IC50s of 243, 453 and 248 nM for HDAC1, HDAC2 and HDAC3, respectively.18) MGCD0103, an orally available HDACi in phase II, inhibited HDAC1 with IC50 of 150 nM, 2- to 10-fold selectivity against HDAC2/3/11.19) Chidamide, a similar structure to MS-275, was approved for treating recurrent and refractory peripheral T-cell lymphoma by China in 2015.9,10) Although a great success appeared in the discovery of new HDAC inhibitors, their deficient clinical efficacy for HDACis against solid tumors is still a problem.20) Hence, the development of selective and broad-spectrum HDAC inhibitors is a research hot.
Indole and its synthetic analogs played a remarkable role in the discovery of antitumor agents as well as novel HDACs inhibitors.15,21–23) One representative indole-based HDACi, panobinostat (Fig. 1), was approved to treat multiple myeloma. Quisinostat, a “second-generation” class I HDACis, showed marked potency toward HDAC1 (IC50 = 0.11 nM) and is currently in clinical studies.24) In our previous work, we discovered that diketopiperazine derivative 2a (Fig. 2) bearing an indole capping group showed moderate HDAC1 inhibition (IC50 = 405 nM) and acceptable selectivity toward other HDAC isozymes.17) In this paper, we replaced the diketopiperazine fragment with a piperazine group and linked it to the indole cap though a carbonyl bridge, and acquired a novel indole-piperazine hybrid skeleton. Here, we reported the design, synthesis and antiproliferative evaluation of these indole-piperazine hybrids as new class I HDACis.

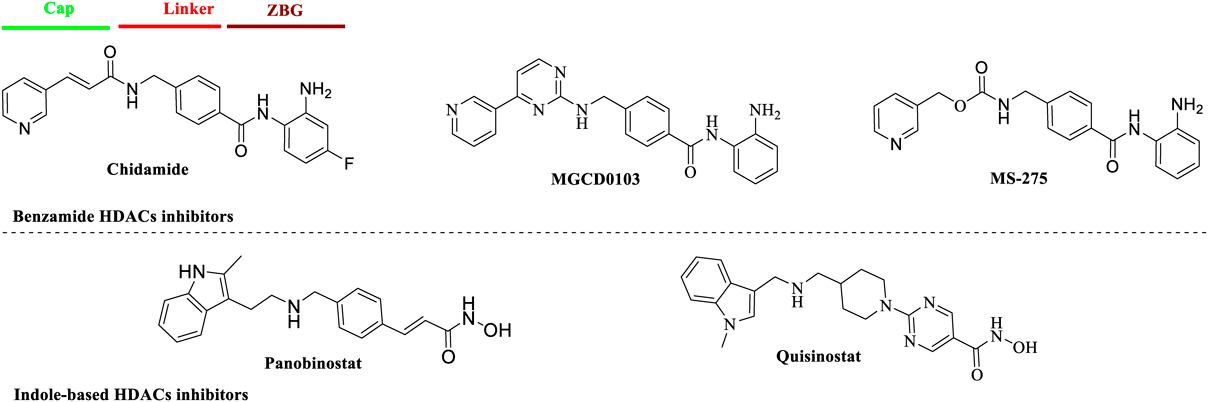
We retained benzamide group as ZBG for its selective binding with class I HDACs and synthesized two attractive indole derivatives 6a and 6b. As shown in Chart 1, Boc-protected piperazine (1) underwent nucleophilic substitution with methyl 4-(bromomethyl)benzoate to give intermediate 2 in acetonitrile. After removal of Boc protecting group, amidation of 3 with indole-3-carboxylic acid or indole-2-carboxylic acid through 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) gave compounds 4a and 4b. Then, the coupling of the hydrolysis product 5a and 5b with o-phenylenediamine yielded desired 6a and 6b.

Reagents and conditions: (a) K2CO3, methyl 4-(bromomethyl)benzoate, acetonitrile, room temperature (r.t.), overnight; (b) TFA, CH2Cl2, reflux, 6 h; (c) Indole-3-carboxylic acid or indole-2-carboxylic acid, HATU, N,N-diisopropylethylamine (DIPEA), N,N-dimethylformamide (DMF), r.t., 6 h. (d) Lithium hydroxide, methanol (MeOH) : H2O = 10 : 1, r.t., overnight; (e) o-phenylenediamine, HATU, DIPEA, DMF, r.t., 6 h.
To study structure and activity relationship (SAR), derivatives with phenyl (8a) or furyl (8b) as capping group was also synthesized. As shown in Chart 2, key intermediate 3 reacted with furan-2-carbonyl chloride or benzoyl chloride gave 7a and 7b. Subsequent hydrolysis and amidation of 7a–7b led to target compounds 8a and 8b.

Reagents and conditions: (a) RCOCl, anhydrous tetrahydrofuran (THF), DIPEA, 0 °C, 4 h; (b) Lithium hydroxide, methanol (MeOH) : H2O = 10 : 1, r.t., overnight; (c) o-Phenylenediamine, HATU, DIPEA, DMF, r.t., 6 h.
According to our previous work,17) substituents on the opposite position of ZBG’s amide had an influence on activity. As exemplified by chidamide, the fluorine atom might improve its lipophilicity and pharmacokinetic properties. Hence, compounds 9a and 9b with fluorine or chlorine substituent were designed and synthesized as shown in Chart 3.
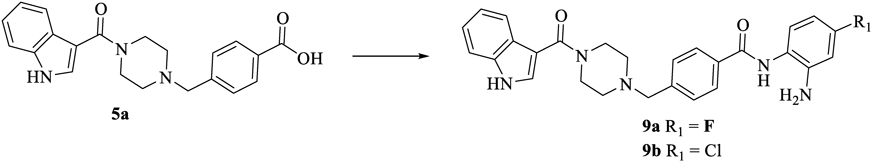
Reagents and conditions: 4-fluorobenzene-1,2-diamine or 4-chlorobenzene-1,2-diamine, HATU, DIPEA, DMF, r.t., 6 h.
All six compounds were preliminarily screened against HDAC1, using chidamide as the positive control. As shown in Table 1, compound 6a exhibited the best inhibitory activity against HDAC1 with IC50 value of 205 nM, a slightly better than that of chidamide (IC50 = 311 nM). Another indole derivative 6b also showed comparable activity (IC50 = 280 nM). The use of phenyl (8a) or furyl (8b) as capping group led to an obvious decrease of enzymatic inhibition (IC50s = 544 and 650 nM, respectively). The introduction of fluorine atom on the para-position of amide (9a) showed a 3-fold decrease against HDAC1 compared with 6a. When a chlorine substituent was applied (9b), the activity was almost lost (IC50 = 1.3 µM). It was clear that the introduction of halogen atoms on ZBG group was unfavorable for HDAC1 activity.
 |
a IC50 values for enzymatic inhibition of HDAC1 enzyme. We ran experiments in duplicate, standard deviation (S.D.) <15%. Assays were performed by Reaction Biology Corporation (Malvern, PA, U.S.A.).
The best compound 6a was tested against other HDAC isozymes including class I HDACs (HDAC2, 3, 8), HDAC 4, 5 (class IIa), and HDAC6 (class IIb) with Chidamide and nonselective approved HDACi SAHA. Besides, the selective class IIa inhibitor TMP269 was also chosen as the positive compound.25) As illustrated in Table 2, 6a showed moderate inhibition toward HDAC2 and HDAC3 with IC50 values of 580 and 630 nM, respectively. The activity against HDAC8 was poor. Besides, 6a had no inhibition of HDAC4–6.
| Compound | IC50a) | ||||||
|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC8 | |
| 6a | 0.205 | 0.580 | 0.630 | >50 | >50 | >50 | 3.550 |
| Chidamide | 0.311 | 0.522 | 0.279 | >50 | >50 | >50 | 1.125 |
| SAHA | 0.004 | 0.011 | 0.003 | >50 | 8.87 | 0.007 | 1.05 |
| TMP269 | >50 | >50 | >50 | 0.133 | 0.090 | NDb) | ND |
a) IC50 values for enzymatic inhibition of HDAC enzymes. We ran experiments in duplicate, S.D. <15%. Assays were performed by Reaction Biology Corporation (Malvern, PA, U.S.A.). b) ND = not determined.
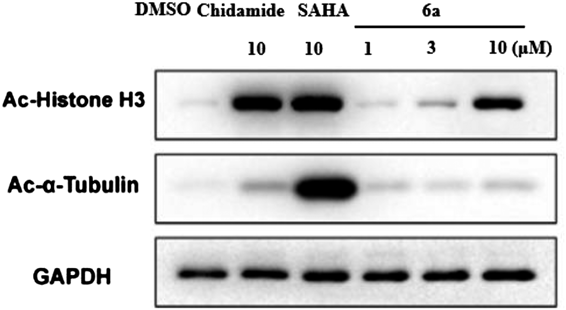
We further validate the selectivity of these hybrids toward class I HDACs by Western blot assay based on the fact that selective inhibition of HDAC1–3 usually causes acetylation of histone H3 without ɑ-tubulin.3) HCT116 cells were treated with representative 6a at 1, 3 and 10 µM for 24 h to measure the acetylation level of H3 acetylation. As shown in Fig. 3, a dose-dependent increase of Ac-H3 was observed. While 6a did not lead to acetylation of ɑ-tubulin compared with nonselective SAHA in this assay.
Molecular docking was performed to rationalize the activity difference between compounds 6a–6b and 8a–8b. The human HDAC1 crystallographic structure (PDB code: 5ICN) was used to simulate the interaction between ligands and the target protein. As shown in Figs. 4A–D, all four compounds could stretch into the narrow active pocket and contact with the Zn ion by a bidentate chelation. Moreover, benzamide ZBG formed three H-bonds with different residues of amino acids such as Gly149, His141 and His140. These interactions ensured their preliminary binding affinity for HDAC1. For compounds 6a (Fig. 4A) or 6b (Fig. 4B), an additional H-bond was observed between the NH atom of indole and the residues (Glu203 for 6a and Asp99 for 6b, respectively) of cap region. This might be responsible for the better HDAC1 inhibition of 6a and 6b. The piperazine and adjacent carbonyl as appropriate linker made the key indole and ZBG part match well with the pocket as shown in the docking results above.
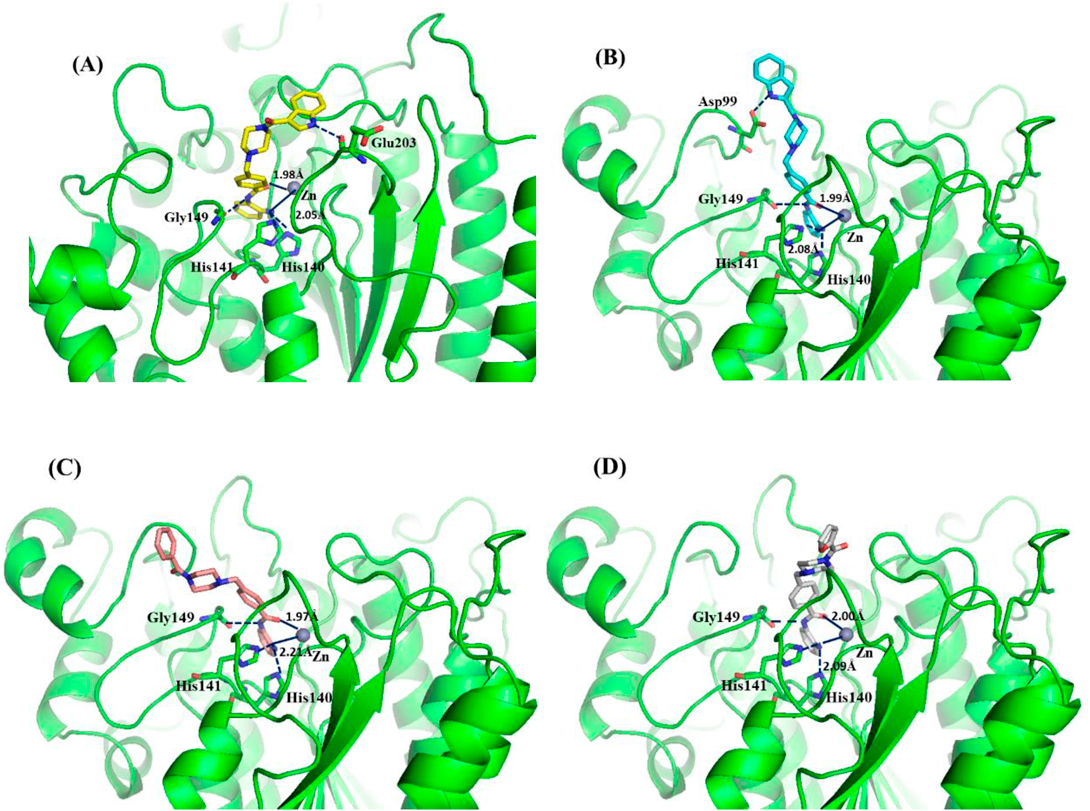
(B) Binding model of 6b (wathet) in the catalytic pocket of HDAC1. (C) Binding model of 8a (light pink) in the catalytic pocket of HDAC1. (D) Binding model of 8b (gray) in the catalytic pocket of HDAC1. Key residues were labeled in green. The hydrogen bonds were labeled in blue. Zinc ion was shown in brown.
Three representative indole derivatives 6a–6b, and 9a were evaluated for their antiproliferative activities against human leukemia cell lines K562 and colon cancer cell line HCT116. As illustrated in Table 3, compound 6a and 6b showed potent antiproliferative activities against K562 and HCT116. Especially for 6a, with IC50s of 3.39 and 2.25 µM, were better than those of chidamide. This trend was almost consistent with their enzymatic activities. Notably, compound 9a also showed acceptable IC50 values in spite of much weaker enzymatic inhibition than 6a, which might be due to its higher lipophilicity.
| Compound | K562 | HCT116 |
|---|---|---|
| 6a | 3.39 | 2.25 |
| 6b | 4.05 | 3.65 |
| 9a | 4.67 | 3.89 |
| Chidamide | 4.54 | 3.32 |
a) IC50 values are averages of three independent experiments, S.D. <10%.
As a result, we designed and obtained a series of aroylpiperazine hybrid derivatives as class I HDACs inhibitors. SAR study showed that indole-based derivative 6a had the best inhibition of HDAC1 with IC50 value of 205 nM, and exhibited a preferred affinity for HDAC1–3 compared with HDAC4–6 or HDAC8. Molecular docking study disclosed that the indole group of 6a had a critical interaction with the residue in cap region of HDAC1 protein. In vitro, 6a showed potent antiproliferative activities, especially for solid tumor cell lines HCT116. Further study for 6a is under way in our lab.
All of the starting reagents were purchased and were used with no additional purification. All of the mentioned yields were for isolated products. Melting points were determined in open capillaries on a WRS-1A digital melting point apparatus (Shenguang). 1H-NMR spectras were detected on a Bruker DRX-400 (400 MHz) using tetramethylsilane (TMS) as internal standard. High resolution mass spectra were obtained from Thermo Scientific Q Exactive. The chemical shifts were reported in ppm (δ) and coupling constants (J) values were given in Hertz (Hz). The purities of all target compounds were tested by HPLC to be >95.0%. HPLC analysis was performed at room temperature using an Agilent Eclipse XDB-C18 (250 × 4.6 mm) and plotted at 254 nm by 30% MeOH/H2O as a mobile phase.
tert-Butyl 4-(4-(Methoxycarbonyl)benzyl)piperazine-1-carboxylate (2)To a stirred mixture of 1 (1 g, 5.37 mmol), potassium carbonate (815 mg, 5.91 mmol) in 30 mL of acetonitrile was added methyl 4-(bromomethyl)benzoate (1.23 g, 5.37 mmol) at 0 °C. After stirring at r.t. for overnight, the reaction mixture was concentrated under reduced pressure. Then the reaction mixture was diluted with saturated sodium chloride (50 mL) and extracted with EtOAc (50 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The white product was obtained by chromatography on a silica gel column in a yield of 90%. 1H-NMR (400 MHz, chloroform-d) δ: 7.99 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 3.91 (s, 3H), 3.56 (s, 2H), 3.44 (s, 4H), 2.39 (s, 4H), 1.45 (s, 9H).
Methyl 4-(Piperazin-1-ylmethyl)benzoate (3)To a solution of 2 (1.71 g, 5.1 mmol) in 50 mL of CH2Cl2 was added TFA (5 mL) at 0 °C. The mixture was refluxed for 6h. After stirring at r.t. for overnight, the reaction mixture was concentrated under reduced pressure. Then water was added (50 mL) and the solution was adjusted to pH = 7 with saturated Na2CO3 solution. The mixture was filtrated and the resulting white solid was washed with water (10 mL). The product was obtained as a white solid, 95% yield. 1H-NMR (400 MHz, methanol-d4) δ: 7.99 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 8.1 Hz, 2H), 3.90 (s, 3H), 3.68 (s, 2H), 3.23 (t, J = 5.2 Hz, 4H), 2.69 (t, J = 5.1 Hz, 4H).
Methyl 4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)benzoate (4a)To a stirred mixture of indole-3-carboxylic acid (427 mg, 2.65 mmol), HATU (936 mg, 2.92 mmol) and DIPEA (1.84 mL, 10.6 mmol) in 20 mL of DMF was added 3 (621 mg, 2.65 mmol) at 0 °C. After stirring at r.t. for 6 h, saturated sodium chloride (100 mL) was added. Then the mixture was extracted with EtOAc (100 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was obtained by chromatography on a silica gel column (652 mg, 65.2%). 1H-NMR (400 MHz, dimethyl sulfoxide (DMSO)-d6) δ: 11.58 (s, 1H), 7.93 (d, J = 8.2 Hz, 2H), 7.69–7.62 (m, 2H), 7.48 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 3.84 (s, 3H), 3.67–3.60 (m, 4H), 3.59 (s, 2H), 2.47–2.35 (m, 4H).
Methyl 4-((4-(1H-Indole-2-carbonyl)piperazin-1-yl)methyl)benzoate (4b)Under the same synthetic condition for 4a, indole-3-carboxylic acid and 3 gave 4a as a white solid, 55.2% yield. 1H-NMR (400 MHz, DMSO-d6) δ: 11.56 (s, 1H), 8.00–7.91 (m, 2H), 7.63–7.55 (m, 1H), 7.55–7.47 (m, 2H), 7.41 (dd, J = 8.3, 1.0 Hz, 1H), 7.18 (m, 1H), 7.04 (m, 1H), 6.78 (dd, J = 2.2, 0.9 Hz, 1H), 3.85 (s, 3H), 3.78 (s, 4H), 3.62 (s, 2H), 2.49–2.40 (m, 4H).
4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)benzoic Acid (5a)To a mixed solution (30 mL) of VMeOH : Vwater = 10 : 1 was added 4a (754.9 mg, 2 mmol) and LiOH (288 mg, 8 mmol). After stirring at r.t. for overnight, the reaction mixture was concentrated under reduced pressure. Additional water (20 mL) was added. Then saturated Na2CO3 solution was added to adjust the PH to neutral. The product was obtained by suction filtration (632.4 mg, 87%). 1H-NMR (400 MHz, DMSO-d6) δ: 11.63 (s, 1H), 7.98 (d, J = 8.2 Hz, 2H), 7.71–7.65 (m, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.2 Hz, 1H), 7.15 (t, J = 7.4 Hz, 1H), 7.10 (t, J = 7.4 Hz, 1H), 3.68–3.61 (m, 4H), 3.59 (s, 2H), 2.46–2.35 (m, 4H).
4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)benzoic Acid (5b)Under the same synthetic condition for 5a, 4b and LiOH gave 5b as an oil, 88.2% yield. Electrospray ionization (ESI)-MS m/z: 364.2 ([M + H]+).
4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)-N-(2-aminophenyl)benzamide (6a)Under the same synthetic condition for 4a, compound 5a and benzene-1,2-diamine gave 6a as a white solid, 63.1% yield. m.p.: 127–128 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 11.60 (s, 1H), 9.66 (s, 1H), 7.95 (d, J = 7.8 Hz, 2H), 7.70–7.63 (m, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.4 Hz, 1H), 7.20–7.05 (m, 3H), 6.97 (t, J = 7.6 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.60 (t, J = 7.5 Hz, 1H), 4.91 (s, 2H), 3.64 (s, 4H), 3.60 (s, 2H), 2.44 (s, 4H). HR-MS (ESI, m/z): Calcd for 454.2238. (C27H28N5O2+ [M + H]+). Found 454.2239.
4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)-N-(2-aminophenyl)benzamide (6b)Under the same synthetic condition for 4a, compound 5b and benzene-1,2-diamine gave 6b as a white solid, 58.2% yield. m.p.: 133–135 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 11.58 (d, J = 2.2 Hz, 1H), 9.67 (s, 1H), 8.07–7.87 (m, 2H), 7.61 (d, J = 8.0 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.24–7.13 (m, 2H), 7.05 (t, J = 7.7 Hz, 1H), 6.98 (td, J = 7.6, 1.6 Hz, 1H), 6.83–6.72 (m, 2H), 6.61 (td, J = 7.5, 1.5 Hz, 1H), 4.91 (s, 2H), 3.79 (s, 4H), 3.62 (s, 2H), 2.48 (dd, J = 6.1, 3.9 Hz, 4H). HR-MS (ESI, m/z): Calcd for 454.2238. (C27H28N5O2+ [M + H]+). Found 454.2245.
Methyl 4-((4-Benzoylpiperazin-1-yl)methyl)benzoate (7a)To a solution of compound 3 (703 mg, 3 mmol) in anhydrous THF (25 mL) was added DIPEA (2.62 mL, 15 mmol). After the mixture was cooled down to 0 °C, benzoyl chloride (348 µL, 3 mmol) was dripped slowly. After stirring for 4 h, the reaction mixture was concentrated under reduced pressure. Additional water (30 mL) was added, and the mixture was extracted with EtOAc (30 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was obtained by chromatography on a silica gel column (746.2 mg, 73.5%) as a light yellow solid. 1H-NMR (400 MHz, methanol-d4) δ: 7.97 (d, J = 8.2 Hz, 2H), 7.52–7.43 (m, 5H), 7.43–7.36 (m, 2H), 3.89 (s, 3H), 3.78 (s, 2H), 3.62 (s, 2H), 3.46 (s, 2H), 2.22 (s, 2H), 2.18 (s, 2H).
Methyl 4-((4-(Furan-2-carbonyl)piperazin-1-yl)methyl)benzoate (7b)Under the same synthetic condition for 7a, compound 3 and furan-2-carbonyl chloride gave 7b as a white solid, 89.5% yield. 1H-NMR (400 MHz, DMSO-d6) δ: 8.03–7.88 (m, 2H), 7.82 (dd, J = 1.8, 0.8 Hz, 1H), 7.54–7.40 (m, 2H), 6.98 (dd, J = 3.5, 0.9 Hz, 1H), 6.61 (dd, J = 3.5, 1.7 Hz, 1H), 3.85 (s, 3H), 3.76–3.62 (m, 4H), 3.60 (s, 2H), 2.43 (t, J = 5.0 Hz, 4H).
N-(2-Aminophenyl)-4-((4-benzoylpiperazin-1-yl)methyl)benzamide (8a)Under the same synthetic condition for 4a, compound 7a and benzene-1, 2-diamine gave 8a as a white solid, 70.4% yield. m.p.: 114–116 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 9.66 (s, 1H), 7.96 (d, J = 8.0 Hz, 2H), 7.49–7.43 (m, 5H), 7.39 (td, J = 4.9, 4.2, 2.8 Hz, 2H), 7.17 (dd, J = 7.9, 1.6 Hz, 1H), 6.98 (td, J = 7.6, 1.6 Hz, 1H), 6.79 (dd, J = 8.0, 1.5 Hz, 1H), 6.61 (td, J = 7.5, 1.5 Hz, 1H), 4.90 (s, 2H), 3.62 (d, J = 17.4 Hz, 4H), 2.48–2.25 (m, 4H). HR-MS (ESI, m/z): Calcd for 415.2129 (C25H27N4O2+ [M + H]+). Found 415.2121.
N-(2-Aminophenyl)-4-((4-(furan-2-carbonyl)piperazin-1-yl)methyl)benzamide (8b)Under the same synthetic condition for 4a, compound 7b and benzene-1,2-diamine gave 8b as a white solid, 77.8% yield. m.p.: 120–122 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 9.66 (s, 1H), 7.97 (d, J = 7.8 Hz, 2H), 7.83 (d, J = 1.4 Hz, 1H), 7.46 (d, J = 8.1 Hz, 2H), 7.17 (dd, J = 7.9, 1.5 Hz, 1H), 7.04–6.92 (m, 2H), 6.79 (dd, J = 8.0, 1.5 Hz, 1H), 6.66–6.54 (m, 2H), 4.91 (s, 2H), 3.68 (s, 4H), 3.61 (s, 2H), 2.45 (t, J = 5.1 Hz, 4H). HR-MS (ESI, m/z): Calcd for 405.1921. (C23H25N4O3+ [M + H]+). Found 405.1925.
4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)-N-(2-amino-4-fluorophenyl)benzamide (9a)Using the synthetic method for 4a, compound 5a and 4-fluorobenzene-1,2-diamine gave 9a as a white solid, 66.7% yield. m.p.: 153–155 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 11.60 (s, 1H), 9.63 (s, 1H), 7.95 (d, J = 7.8 Hz, 2H), 7.72–7.60 (m, 2H), 7.46 (d, J = 8.0 Hz, 3H), 7.43 (d, J = 8.0 Hz, 2H), 7.18–7.06 (m, 3H), 6.80 (s, 1H), 6.59 (d, J = 8.3 Hz, 1H), 5.25 (s, 1H), 3.64 (s, 4H), 3.61 (s, 2H), 2.45 (s, 4H). HR-MS (ESI, m/z): Calcd for 472.2143. (C27H27FN5O2+ [M + H]+). Found 472.2144.
4-((4-(1H-Indole-3-carbonyl)piperazin-1-yl)methyl)-N-(2-amino-4-chlorophenyl)benzamide (9b)Using the synthetic method for 4a, compound 5a and 4-chlorobenzene-1, 2-diamine gave 9b as a white solid, 70.5% yield. m.p.: 124–125 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 11.58 (s, 1H), 9.57 (s, 1H), 7.95 (d, J = 7.9 Hz, 2H), 7.70–7.63 (m, 2H), 7.49–7.40 (m, 3H), 7.18–7.05 (m, 3H), 6.54 (dd, J = 11.2, 2.9 Hz, 1H), 6.35 (td, J = 8.5, 2.8 Hz, 1H), 5.22 (s, 2H), 3.64 (s, 4H), 3.60 (s, 2H), 2.44 (s, 4H). HR-MS (ESI, m/z): Calcd for 488.1848. (C27H2735ClN5O2+ [M + H]+). Found 488.1844. Calcd for 490.1818. (C27H2737ClN5O2+ [M + H]+). Found 490.1828.
In vitro HDAC Enzyme AssayIC50 testing of compounds were performed by Reaction Biology Corporation. The HDACs were isolated from a baculovirus expression system in Sf9 cells using an acetylated fluorogenic peptide RHKKAc as substrate. The reaction buffer was made up of 50 mM Tris–HCl pH 8.0, 127 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA, and a final concentration of 1% DMSO. Compounds were delivered in DMSO and delivered to enzyme mixture with preincubation of 5–10 min followed by substrate addition and incubation for 2 h at 30 °C. Trichostatin A and a developer were added to quench the reaction and generate fluorescence, respectively. Dose–response curves were generated starting at 30 µM compound with 3-fold serial dilutions to generate a 10-dose plot. IC50 values were then generated from the resulting plots.
Cell Culture and Antiproliferative AssayThe cells were cultured in IMDM medium with 20% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin. All cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 in air. Briefly, 100 µL cell suspension or completed medium were plated into 96-well plate. Compounds were added and incubated for 72 h; Then, 22 µL Alamar blue solution (1 mM) were pipetted into each well of 96-well plate; and the plate was incubated for an additional 5–6 h. The absorbance (optical density (OD)) was read at 530/590 nm. Data were normalized to vehicle groups (DMSO) and represented as the means of three independent measurements with standard errors of <20%. The IC50 values were calculated using Prism 5.0.
Western Blotting AssayHCT116 cells (1 × 106) were seeded overnight and incubated with compound 6a for 24 h on indicated concentrations. Cell extract was prepared by lysing cultured cells with a mammalian protein extraction reagent supplemented with ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor for 15 min. Supernatants were collected following centrifugation of lysed cells at 15000 × g for 10 min at 4 °C. Then, 30 mg of total protein per sample was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane to analyze the cell lysate. Membranes with immobilized proteins were probed with antibodies for Ac-H3 (abcom, AB32129) and Ac-ɑ-tubulin (Cell Signaling, 2144). The reactive protein bands were visualized using an ECL detection system.
Computational MethodsDiscovery Studio 3.0 software (BIOVIA, 5005 Wateridge Vista Drive, San Diego, CA92121 U.S.A.) was used for simulation. Docking was conducted using cdocker based on the cocrystal of HDAC1 (PDB: 5ICN). HDAC1 was used as the receptor. The cavity occupied by a peptide inhibitor was selected as binding site. The docking sphere radius value based on the peptide inhibitor was default. Water molecules outside the binding pocket were excluded. The energy minimization for compound 6a–6b, 8a–8b were performed by Powell's method for 1000 iterations using Tripos force field and with Gasteiger-Hückel charge. The other docking parameters were kept as default.
This work was supported by the National Natural Science Foundation of China (81903464), Guangxi First-class Disciplines (Agricultural Resources and Environment) and the Natural Science Basic Research Plan in Shaanxi Province of China (2020JQ-284).
The authors declare no conflict of interest.
This article contains supplementary materials.