Regular Articles
Synthesis and Characterization of Novel Radioiodinated Triazole-Pyrolidine Derivative to Detect Orexin 2 Receptor in the Brain
2023 年 71 巻 3 号 p. 234-239
詳細
2023 年 71 巻 3 号 p. 234-239
It is generally accepted that the orexin 2 receptor (OX2R) plays a critical role in the arousal-promoting function, and in vivo imaging of OX2R is expected to contribute to elucidation of orexin systems and the development of drugs to treat sleep disorder. In this study, we newly synthesized and characterized a radioiodinated triazole-pyrolidine derivative ([125I]TPI) to detect OX2R in the brain. In vitro studies using OX1R or OX2R expression cells showed selective binding of [125I]TPI to OX2R. In addition, in vitro autoradiography using rat brain sections showed high accumulation of radioactivity in the OX2R expression region. However, [125I]TPI showed low brain uptake in normal mice. These results suggest that [125I]TPI has a fundamental character to detect OX2R in vitro, but further structural modification to improve brain pharmacokinetics is required to use it for in vivo detection of OX2R.
The orexin system is made up of two G-protein coupled receptors, orexin 1 receptor (OX1R) and orexin 2 receptor, (OX2R), and two hypothalamic neuropeptides, orexin A and orexin B.1,2) The orexin system is involved in diverse physiological functions, such as the sleep-wake cycle, emotional behavior, reward-seeking, and feeding.2) Additionally, recent research suggested the involvement of the orexin system in neurodegenerative diseases including Alzheimer's disease and Parkinson's disease.3,4) Both OX1R and OX2R are widely expressed throughout the central nervous system (CNS) including cerebral cortex, hippocampus, and thalamus, but these show partially different distributions. For example, OX1R is selectively and strongly expressed in the locus coeruleus, whereas the selective expression of OX2R is observed in the tuberomammillary nucleus, nucleus accumbens, and paraventricular nucleus. These suggest that OX1R and OX2R are involved in different physiological functions.2,5,6) Especially, since OX2R mainly affects sleep,2) many antagonists targeting OX2R for the treatment of sleep disorders have been developed. In addition, some agonists targeting OX2R have also been developed to treat narcolepsy.7,8) However, the relationships between the function or various diseases and orexin receptors have not been fully elucidated.
Nuclear medicine techniques including positron emission tomography (PET) and single photon emission computed tomography (SPECT) have the ability to detect and quantify molecular targets within CNS.9,10) Since non-invasive imaging of OX2R could be useful for not only elucidation of orexin systems but also development of agonists/antagonists, several PET probes targeting OX2R have been developed.11–17) First, 11C-labeled probes, such as [11C]EMPA, [11C]BBAC, and [11C]CW4, were reported, but these showed insufficient brain uptake or high non-specific accumulation in the brain for in vivo imaging. More recently, two 18F-labeled probes, [18F]DAN-1 and [18F]seltorexant, were also reported. [18F]DAN-1 showed moderate uptake into the mouse brain. Moreover, the radioactivity in the brain was reduced by co-injection with an excess amount of non-radioactive DAN-1, suggesting the binding of DAN-1 to OX2R in the living mouse brain. However, high non-specific accumulation was observed in the brain.16) [18F]Seltorexant also penetrates the blood-brain barrier, but since it functions as a substrate of P-glycoprotein, an increase in brain uptake was observed in an in vivo blocking study.17) Taken together, no promising probes for in vivo imaging of OX2R have been developed so far.
To develop novel OX2R imaging probes, we focused on the triazole-pyrolidine (TP) scaffold which is one of the backbones of orexin receptor antagonists. TP derivatives showed brain uptake, and a structure–activity relationship study suggested that (S)-(2-(5-(3-chloro-2-methylphenyl)-4H-1,2,4-triazol-3-yl)pyrrolidin-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (TPCl) showed high selectivity for OX2R over OX1R18) (Fig. 1). Since iodine has several radioisotopes, including I-123 (SPECT), I-124 (PET), and I-125 (basic research), we newly designed a TP derivative (TPI), replacing a chloride atom of TPCl with an iodine atom, to detect OX2R in the brain (Fig. 1).

All reagents were obtained commercially and used without further purification unless otherwise indicated. 1H- and 13C-NMR spectra were obtained on JEOL JNM-ECS400 (JEOL, Tokyo, Japan) with tetramethylsilane as an internal standard. Coupling constants are reported in hertz. Multiplicity was defined by s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra (MS) and high-resolution MS (HRMS) were obtained on SHIMADZU LCMS-2020 EV and LCMS-IT-TOF ((Shimadzu, Kyoto, Japan), respectively. Reversed-phase HPLC was performed with a Shimadzu system (LC-20AD pump with SPD-20A UV detector, λ = 254 nm) using a Cosmosil C18 column (5C18-AR-II 4.6 mm I.D. × 150 mm, Nacalai Tesque, Kyoto, Japan).
Chemistry3-Bromo-2-methylbenzohydrazide (1)2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) (38.5 mg, 0.12 mmol) was added to a solution of 3-bromo-2-methylbenzoic acid (21.5 mg, 0.1 mmol) and diisopropylethylamine (DIPEA) (43 µL, 0.27 mmol) in dimethylformamide (DMF) (5 mL). The solution was stirred at room temperature for 15 min, and then the mixture was cooled to 0 °C before hydrazine (350 µL, 0.35 mmol) was added. The solution was stirred at room temperature for 20 min, and then the mixture was diluted with CHCl3 and washed with sat. aq. NaHCO3. The extracted solution was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (chloroform/methanol = 20/1) to give 1 as a white solid (19.7 mg, 86%). 1H-NMR (400 MHz, CDCl3) δ: 7.64 (dd, J = 8.1, 1.2 Hz, 1H), 7.09 (t, J = 8.1 Hz, 1H), 6.94 (s, 1H), 4.12 (s, 2H), 2.47 (s, 3H). MS (electrospray ionization (ESI)) m/z 229.0 [MH+].
3-Iodo-2-methylbenzohydrazide (2)Compound 2 was synthesized from 3-iodo-2-chloro-methylbenzoic acid according to the method described above to prepare 1 (214 mg, 77%). 1H-NMR (400 MHz, CDCl3) δ: 7.91 (d, J = 8.1 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 4.33 (s, 2H), 2.43 (s, 3H). MS (ESI) m/z 276.9 [MH+].
tert-Butyl (S)-2-(5-(3-Bromo-2-methylphenyl)-4H-1,2,4-triazol-3-yl)pyrrolidine-1-carboxylate (3)K2CO3 (88 mg, 0.63 mmol) was added to a solution of 1 (145 mg, 0.63 mmol) and (S)-1-boc-2-cyanopyrrolidine (232 mg, 1.14 mmol) in n-BuOH (10 mL) and the mixture was refluxed (130 °C) for 16 h. The mixture was concentrated in vacuo. To residue was added CHCl3, and then the mixture was acidified with HCl aq. (pH 5). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (ethyl acetate/hexane = 2/3) to give 3 as a white solid (195 mg, 75%). 1H-NMR (400 MHz, CDCl3) δ: 12.24 (s, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.08 (t, J = 7.8 Hz, 1H), 5.04 (q, J = 2.9 Hz, 1H), 4.12 (q, J = 7.2 Hz, 2H), 2.61 (s, 3H), 2.23–2.14 (m, 2H), 1.98 (s, 2H), 1.48 (s, 9H). MS (ESI) m/z 407.1 [MH+].
tert-Butyl (S)-2-(5-(3-Iodo-2-methylphenyl)-4H-1,2,4-triazol-3-yl)pyrrolidine-1-carboxylate (4)Compound 4 was synthesized from 2 according to the method described above to prepare 3 (337 mg, 23%). 1H-NMR (400 MHz, CDCl3) δ: 7.82 (d, J = 7.5 Hz, 1H), 7.65 (d, J = 7.5 Hz, 1H), 6.86 (t, J = 7.5 Hz, 1H), 4.95 (dd, J = 8.1, 2.9 Hz, 1H), 4.05 (q, J = 7.0 Hz, 1H), 3.36–3.33 (m, 1H), 2.80 (s, 1H), 2.60 (s, 3H), 2.15 (s, 1H), 2.02 (s, 1H), 1.98 (s, 1H), 1.95–1.92 (m, 1H), 1.43 (s, 9H). MS (ESI) m/z 455.0 [MH+].
(S)-3-(3-Bromo-2-methylphenyl)-5-(pyrrolidin-2-yl)-4H-1,2,4-triazole (5)Trifluoroacetic acid (TFA) (5 mL) was added to a solution of 3 (1069 mg, 2.62 mmol) in CH2Cl2 (5 mL). The mixture was stirred at room temperature for 1 h. The mixture was neutralized with NaHCO3 and extracted with CHCl3. The extracted solution was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (chloroform/methanol = 1/1) to give 5 (491 mg, 61%). 1H-NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 7.2 Hz, 1H), 4.53 (d, J = 6.4 Hz, 1H), 3.11 (t, J = 6.7 Hz, 2H), 2.59 (s, 4H), 2.32 (q, J = 6.4 Hz, 1H), 2.10 (q, J = 7.2 Hz, 1H), 1.89 (t, J = 3.2 Hz, 2H). MS (ESI) m/z 307.1 [MH+].
(S)-3-(3-Iodo-2-methylphenyl)-5-(pyrrolidin-2-yl)-4H-1,2,4-triazole (6)Compound 6 was synthesized from 4 according to the method described above to prepare 5 (135 mg, 52%). 1H-NMR (400 MHz, CDCl3) δ: 7.79 (d, J = 7.5 Hz, 1H), 7.49 (d, J = 7.5 Hz, 1H), 6.78 (t, J = 8.1 Hz, 1H), 4.37 (t, J = 7.5 Hz, 1H), 3.06–2.92 (m, 2H), 2.51 (s, 3H), 2.20–2.13 (m, 1H), 2.00–1.93 (m, 2H), 1.83–1.76 (m, 2H). MS (ESI) m/z 354.9 [MH+].
(S)-(2-(5-(3-Bromo-2-methylphenyl)-4H-1,2,4-triazol-3-yl)pyrrolidin-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (7)TBTU (617 mg, 1.91 mmol) was added to a solution of 5-methyl-2-[1,2,3]triazole-2-yl-benzoic acid (325 mg, 1.59 mmol) and DIPEA (272 µL, 1.91 mmol) in CH2Cl2 (5 mL). The mixture was stirred at room temperature for 10 min, before 5 in CH2Cl2 (5 mL) was added and stirred at room temperature for 1 h. The mixture was diluted with CHCl3 and washed with sat. aq. NaHCO3. The extracted solution was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography (ethyl acetate/hexane = 1/1) to give 7 (645 mg, 82%). 1H-NMR (400 MHz, CDCl3) δ: 12.35 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.79–7.75 (m, 3H), 7.61 (d, J = 7.0 Hz, 1H), 7.38 (dd, J = 8.4, 1.2 Hz, 2H), 7.13 (t, J = 7.8 Hz, 1H), 5.51 (s, 1H), 3.13 (s, 1H), 2.80 (s, 3H), 2.68 (s, 3H), 1.97 (s, 2H), 1.34–1.25 (m, 2H). MS (ESI) m/z 492.2 [MH+].
(S)-(2-(5-(3-Iodo-2-methylphenyl)-4H-1,2,4-triazol-3-yl)pyrrolidin-1-yl)(5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone (8:TPI)Compound 8 was synthesized from 6 according to the method described above to prepare 7 (95 mg, 47%). 1H-NMR (400 MHz, CDCl3) δ: 7.85 (d, J = 8.7 Hz, 1H), 7.80 (d, J = 7.5 Hz, 1H), 7.66 (s, 2H), 7.61 (d, J = 6.4 Hz, 1H), 7.27 (d, J = 8.1 Hz, 1H), 6.84 (t, J = 8.1 Hz, 1H), 5.41 (s, 1H), 4.27 (q, J = 7.0 Hz, 1H), 4.03 (q, J = 7.5 Hz, 1H), 2.58 (s, 3H), 2.33 (s, 3H), 2.07 (s, 2H), 1.95 (s, 1H), 1.26 (t, J = 7.0 Hz, 1H), 1.16 (t, J = 7.0 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ: 139.891, 138.613, 135.801, 133.360, 131.890, 131.100, 130.213, 128.144, 127.191, 122.052, 103.726, 53.297, 48.615, 29.221, 26.695, 24.540, 20.917. HRMS (ESI) m/z Calcd for C23H22IN7O 540.1009. Found 540.0635.
(S)-(5-Methyl-2-(2H-1,2,3-triazol-2-yl)phenyl)(2-(5-(2-methyl-3-(tributylatannyl)phenyl)-4H-1,2,4-triazol-3-yl)pyrrolodin-1-yl)methanone (9)A mixture of 7 (108 mg, 0.22 mmol), Pd(PPh3)4 (201 mg, 0.17 mmol), and bis(tributyltin) (435 µL, 0.87 mmol) in a mixed solvent (2 mL, dioxan/Et3N = 3/1) was stirred at 105 °C for 4 h. The solvent was removed, and the residue was purified with silica gel chromatography (ethyl acetate/hexane = 5/1) to give 9 (89 mg, 58%). 1H-NMR (400 MHz, CDCl3) δ: 12.07 (s, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.77–7.75 (m, 3H), 7.43 (d, J = 7.2 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 5.51 (s, 1H), 3.27 (s, 1H), 3.11 (s, 1H), 2.63 (s, 3H), 2.44 (s, 3H), 2.30 (d, J = 9.9 Hz, 2H), 2.00–1.95 (m, 2H), 1.57–1.50 (m, 5H), 1.35 (q, J = 7.5 Hz, 9H), 1.10 (t, J = 8.1 Hz, 6H), 0.89 (t, J = 7.2 Hz, 9H). MS (ESI) m/z 702.3 [MH+].
RadiosynthesisTo initiate the reaction, compound 9 (100 µg/100 µL EtOH) was added to a mixture of [125I]NaI (3.7 MBq, molar activity 81.4 TBq mmol−1), 100 µL of H2O2 aq. (3%), and 100 µL of 1 N HCl aq. in a sealed vial. The reaction was allowed to proceed at room temperature for 10 min and terminated by the addition of saturated NaHSO3 aq. (200 µL). After neutralization with sodium hydrogen carbonate and extraction with ethyl acetate, the extract was dried by passing through an anhydrous Na2SO4 column. The solution was blown dry with a stream of nitrogen gas. The radioiodinated ligand was purified by HPLC on a Cosmosil C18 column with a solvent of acetonitrile/H2O = 10/90 (0 min) to 100/0 (10 min) at a flow rate of 1.0 mL/min.
Cell CultureChinese hamster ovary (CHO)-K1 cells stably expressing human OX1R (CHO-OX1R) or OX2R (CHO-OX2R) were purchased from GeneScript (New Jersey, U.S.A.). Cells were maintained in Ham’s F 12 medium (Nacalai Tesque) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific, MA, U.S.A.) and 50 µg/mL G418 in an atmosphere containing 5% CO2 at 37 °C.
Cellular Binding StudyCHO-OX1R or CHO-OX2R cells were seeded in a 6-well-plate (1 × 106 cells per well) and incubated in an atmosphere containing 5% CO2 at 37 °C and then at 37 °C for 24 h. After incubation, the cells were treated with 2 mL of [125I]TPI (26.5 kBq/well) at room temperature for 1.5 h. Then, the medium was refreshed, and the cells were further incubated for 1 h. The wells were washed with 1 mL of 1% bovine serum albumin (BSA) and 1% dimethyl sulfoxide (DMSO)/phosphate buffer saline (PBS) (×3), and dissolved with 2 mL of 1 N NaOH. The cell lysis solutions were poured into tubes. The radioactivity of each tube was measured using an automatic γ-counter. For blocking, cells were treated with 1 mL of [125I]TPI (26.5 kBq/well) and 1 mL of almorexant (dual orexin receptors (OXR) antagonist19)) solution (5 µM) at room temperature for 1.5 h. The protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific).
In Vitro Saturation Binding AssayCHO-OX2R cells were seeded in a 12-well-plate (4 × 105 cells per well) and then incubated at 37 °C in an atmosphere containing 5% CO2 for 24 h. After removing the medium, a mixture containing radioactive and nonradioactive compounds ([125I]TPI and TPI, final concentrations: 0–800 nM, 0.04–46 kBq/mL, respectively) in Ham’s F 12 medium (0.6 mL) was added to each well, and plates were incubated at 37 °C for 1.5 h. Non-specific binding was assessed in the presence of almorexant (5 µM). Then, the medium was refreshed, and the cells were further incubated for 1 h. After incubation, the wells were washed with 1 mL of 1% BSA and 1% DMSO/PBS (×3), and dissolved with 1 N NaOH (0.6 mL×2). The cell lysis solutions were poured into tubes. The radioactivity of each tube was measured using an automatic γ-counter. Dissociation constant (Kd) values were determined by Scatchard analysis using GraphPad Prism software (version 6.0; GraphPad Software, CA, U.S.A.).
AnimalsAll animal experiments were conducted in accordance with our institutional guidelines and approved by Kyoto University. CD Sprague-Dawley rats and ddY mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). The animals were housed in a sterile environment under a 12-h light–dark cycle, fed standard chow, and had free access to water. All efforts were made to minimize suffering.
In Vitro AutoradiographyCD Sprague-Dawley rat (male, 10 weeks old) brain sections (30 µM thick) were used in this study. Each section was incubated with a 2% DMSO solution of [125I]TPI (22.2 kBq/mL) at room temperature for 30 min. For blocking experiments, adjacent sections were incubated with a 2% DMSO solution of [125I]TPI (22.2 kBq/mL) in the presence of almorexant (10 µM). The sections were washed with 50% EtOH (1 min×4). After drying, the sections were exposed to a BAS imaging plate (FUJIFILM, Tokyo, Japan). Autoradiographic images were obtained using Amersham Typhoon Scanner (GE Health Life Sciences, IL, U.S.A.).
BiodistributionA biodistribution study was conducted using ddY mice (male, 5 weeks old). [125I]TPI (25.7 kBq/100 µL in 10% EtOH containing 0.1% Tween 80) was administered to mice by tail-vein injection. At 2, 10, 30, and 60 min after administration, mice were sacrificed. Radioactivity in blood and organs was measured using an automatic γ-counter.
Statistical AnalysisStatistical analysis was performed using one-way ANOVA followed by Tukey’s test using GraphPad Prism software.
The synthetic route of TPI is shown in Chart 1. First, we synthesized a hydrazine derivative (1, 2), and then reacted it with (S)-1-boc-2-cyanopyrrolidine in the presence of K2CO3 to obtain 3 and 4. After deprotection of the tert-butoxycarbonyl group with TFA, amide coupling with 5-methyl-2-[1,2,3]triazole-2-yl-benzoic acid yielded 7 and 8 (TPI). The tributyltin compound (9) that serves as the precursor of the 125I-labeling reaction was synthesized from a corresponding bromo compound (7).

As shown in Chart 2, [125I]TPI was synthesized by an iododestannylation reaction with [125I]NaI, hydrogen peroxide, and hydrochloric acid. [125I]TPI was purified by HPLC after the reaction was completed. The radiochemical yield and radiochemical purity of [125I]TPI were 67.3% and >95%, respectively.

First, we investigated the cellular binding of [125I]TPI with OX1R or OX2R expression cells (Fig. 2). [125I]TPI showed 3.56 times higher binding to OX2R expression cells (8.62% dose/mg protein) than that to OX1R expression cells (2.42% dose/mg protein). In addition, the significant blocking of [125I]TPI induced by almorexant, a dual OXR antagonist,19) was only observed in OX2R expression cells (OX1R expression cells: 1.31% dose/mg protein, OX2R expression cells: 1.27% dose/mg protein). These results suggested that [125I]TPI can selectively bind to OX2R in vitro.
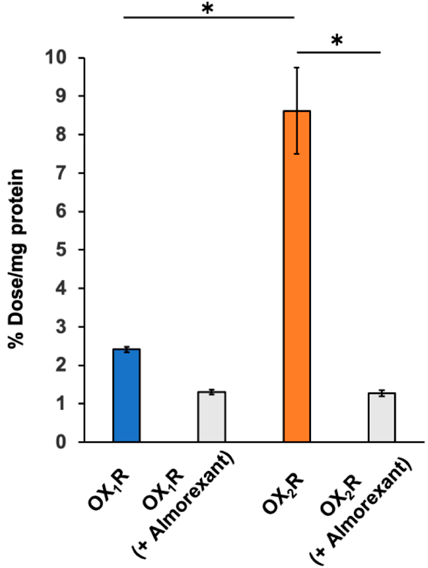
n = 3, * p < 0.05.
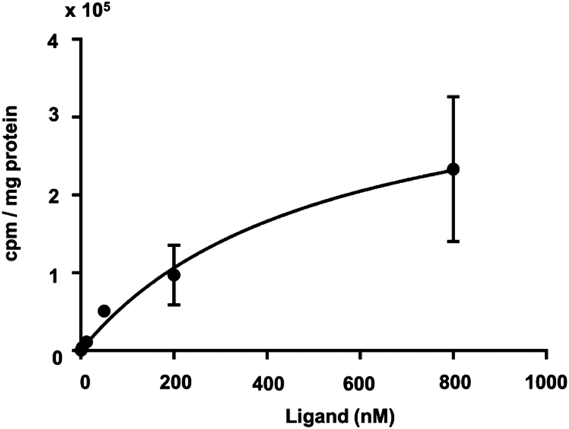
Subsequently, a binding saturation assay was performed to quantitatively evaluate the binding affinity of [125I]TPI for OX2R expression cells (Fig. 3). The Kd value of [125I]TPI was determined to be 520 nM, suggesting that [125I]TPI has moderate affinity for OX2R. However, [125I]TPI showed much lower binding than [3H]EMPA (Kd = 2.7 nM), which is a 3H-labeled compound of the PET probe targeting OX2R reported previously.13,20) We used CHO-OX2R cells, but the Kd value of [3H]EMPA was determined with HEK293 cells expressing human OX2R. This may have affected the differences in binding affinity of these compounds, since the results of binding studies generally depend on the amount and state of OX2R expressed on the membrane surface. A previous report indicated that TPCl, a lead compound of TPI, has high inhibitory activity for OX2R (IC50 = 29 nM).18) This value does not indicate direct binding affinity, but conversion from the chlorine to iodine atom may decrease the binding affinity for OX2R.
We performed an in vitro autoradiographic study with normal rat brain sections to evaluate the distribution of [125I]TPI (Fig. 4). Radioactivity accumulation was observed in some regions, which were known to express OX2R.6) The distribution pattern was consistent with that of [3H]EMPA.12,20,21) In addition, the radioactivity of [125I]TPI was markedly decreased by the addition of an excess amount of almorexant. In the cellular binding study, the binding of [125I]TPI to OX1R expression cells was inhibited by approximately 45% by the addition of almorexant. Although this was not significant, the radioactivity observed in the autoradiographic image may be influenced by [125I]TPI bound to OX1R. Some radioactivity was not completely blocked, which may have been due to non-specific binding caused by the high lipophilicity of [125I]TPI. These results suggest that [125I]TPI has sufficient affinity for detecting OX2R in rat brain sections in vitro, although [125I]TPI showed a high Kd value (520 nM).
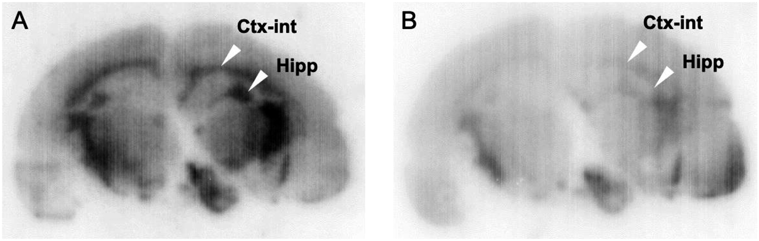
A shows total binding ([125I]TPI), B shows non-specific binding ([125I]TPI + Almorexant).
Finally, a biodistribution study of [125I]TPI was carried out using normal mice (Table 1). The ideal brain pharmacokinetics of an OX2R imaging probe would be for it to exhibit high uptake early post injection and rapid clearance with time, while it is partially retained due to binding to OX2R. [125I]TPI showed low brain uptake at 2 min postinjection (0.55% injected dose (ID)/g), and rapid clearance with time (0.08% ID/g), suggesting that the brain pharmacokinetics of [125I]TPI may not be sufficient for in vivo imaging of OX2R. In general, a low molecular weight and moderate lipophilicity are desirable for CNS imaging probes.9,10) The unfavorable brain pharmacokinetics of [125I]TPI may be caused by a relatively large molecular weight (537 Da) and high lipophilicity (clog P = 3.1, calculated by ChemDraw professional 17.0).
| Tissue | Time after injection (min) | |||
|---|---|---|---|---|
| 2 | 10 | 30 | 60 | |
| Blood | 3.87 ± 0.42 | 2.64 ± 0.44 | 1.79 ± 0.25 | 1.54 ± 0.16 |
| Spleen | 2.51 ± 0.34 | 2.09 ± 0.23 | 1.02 ± 0.12 | 0.83 ± 0.07 |
| Pancreas | 5.47 ± 0.63 | 3.32 ± 0.33 | 1.12 ± 0.20 | 0.63 ± 0.04 |
| Stomachb) | 1.04 ± 0.07 | 2.99 ± 0.45 | 6.08 ± 0.66 | 7.58 ± 2.12 |
| Intestine | 2.82 ± 0.44 | 8.87 ± 1.34 | 15.11 ± 2.67 | 22.73 ± 3.04 |
| Kidney | 10.49 ± 1.23 | 3.98 ± 0.51 | 1.62 ± 0.17 | 1.23 ± 0.18 |
| Liver | 24.97 ± 2.92 | 32.41 ± 3.29 | 25.18 ± 3.37 | 13.59 ± 0.93 |
| Heart | 7.99 ± 1.07 | 2.46 ± 0.33 | 1.04 ± 0.19 | 0.74 ± 0.12 |
| Lung | 6.17 ± 0.50 | 2.82 ± 0.55 | 1.63 ± 0.33 | 1.35 ± 0.21 |
| Brain | 0.55 ± 0.06 | 0.44 ± 0.11 | 0.13 ± 0.03 | 0.08 ± 0.06 |
| Thyroidb) | 0.10 ± 0.02 | 0.10 ± 0.04 | 0.07 ± 0.08 | 0.06 ± 0.04 |
a) Expressed as % injected dose per gram. Each value represents the mean ± standard deviation (S.D.) for 5 animals. b) Expressed as % injected dose per organ.
Next, we also evaluated the whole-body distribution of radioactivity. Radioactivity after injection of [125I]TPI into normal mice mainly accumulated in the liver (32.4% ID/g at 10 min after intravenous injection). Thereafter, the radioactivity was translocated into the intestine (22.7% ID/g at 60 min after intravenous injection). These distributions are typical of lipophilic low-molecular-wight compounds. No marked uptake in the thyroid was observed (0.06% ID at 60 min after intravenous injection), suggesting that [125I]TPI may be stable for deiodination metabolism by 60 min after intravenous injection.
We developed a novel radioiodinated probe, [125I]TPI, to detect OX2R in the brain. In vitro studies using OXR expression cells indicated that [125I]TPI selectively bound to OX2R. In addition, [125I]TPI could detect OX2R in rat brain sections. However, the brain uptake was too low to perform in vivo imaging. These results suggest that an additional structure–activity relationship study based on the TP scaffold to enhance brain uptake may lead to the development of useful OX2R imaging probes.
This research was supported by JSPS KAKENHI Grant Numbers 19H05018 and 21H02869.
The authors declare no conflict of interest.