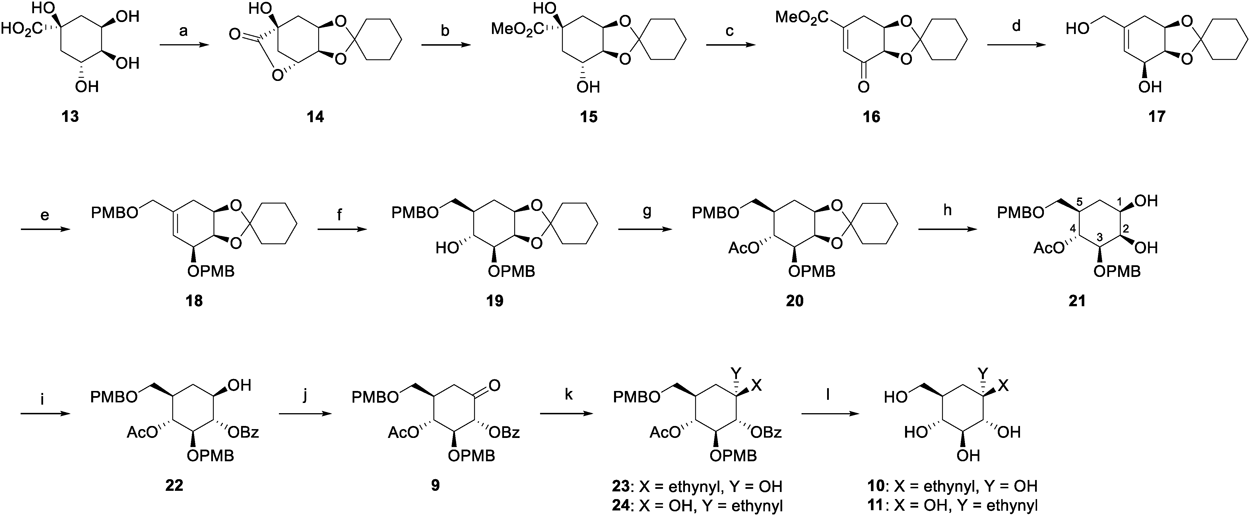
Experimental
GeneralAll reactions were carried out under an argon atmosphere, unless otherwise noted. All reagents and solvents were purchased from commercial vendors and used without further purification, unless indicated otherwise. Pyridine was distilled over CaH2 and stored over activated molecular sieves 4 Å (MS4A). 1H- and 13C-NMR spectra were recorded on a JEOL JNM AL-400 spectrometer or JNM ECS-400 spectrometer (400 MHz for 1H-NMR and 100 MHz for 13C-NMR). Chemical shifts (δ) were expressed in parts per million and internally referenced (7.26 ppm for CDCl3, 3.30 ppm for CD3OD or 2.49 ppm for dimethyl sulfoxide (DMSO)-d6 for 1H-NMR, 77.0 ppm for CDCl3, 49.0 ppm for CD3OD). Electrospray ionization (ESI) mass spectra were taken on a JMS T100LP instrument or a Waters Xevo Q-Tof mass spectrometer. Flash column chromatography was performed using silica gel 60N [spherical neutral (63–210 µm)] from Kanto Chemical Co., Inc. (Tokyo, Japan) or silica gel PSQ 100B (63–210 µm) from Fuji Silysia Chemical Co., Ltd. (Kasugai, Aichi, Japan).
3,4-O-Cyclohexylidenequinic Acid-1,5-lactone (14)23)Cyclohexanone (9.60 mL, 93.6 mmol) was added dropwise to a suspension of D-quinic acid (13) (12.0 g, 62.4 mmol) and p-toluenesulfonic acid monohydrate (544 mg, 3.16 mmol) in toluene (120 mL), and then the mixture was azeotoropic-refluxed with a Dean-Stark apparatus for 20 h. After the reaction mixture was cooled to room temperature (r.t.), the reaction mixture was partitioned between EtOAc and H2O. The organic layer was washed with saturated NaHCO3 aqueous solution, H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. n-Hexane was added to the residue, and the resulting precipitate was collected by filtration, washed with n-hexane to give 14 as a colorless solid (14.9 g, 94%). 1H-NMR (CDCl3) δ: 4.74 (1H, dd, J = 6.0 Hz, 2.5 Hz), 4.48 (1H, ddd, J = 7.9 Hz, 6.5 Hz, 3.2 Hz), 4.31 (1H, ddd, J = 6.5 Hz, 2.5 Hz, 1.3 Hz), 2.67 (1H, d, J = 11.4 Hz), 2.67 (1H, s), 2.35 (1H, ddd, J = 14.7 Hz, 7.9 Hz, 2.3 Hz), 2.29 (1H, dddd, J = 11.4 Hz, 6.0 Hz, 2.3 Hz, 1.3 Hz), 2.19 (1H, dd, J = 14.7 Hz, 3.2 Hz), 1.70–1.73 (2H, m), 1.62–1.68 (2H, m), 1.52–1.60 (4H, m), 1.37–1.43 (2H, m); 13C-NMR (CDCl3) δ: 179.0, 110.6, 75.9, 71.7, 71.5, 71.0, 38.3, 36.8, 34.3, 33.6, 25.0, 23.9, 23.4; high resolution (HR)-MS (ESI) m/z: 255.1209 [M + H]+ (Calcd for C13H19O5: 255.1232).
Methyl 3,4-O-Cyclohexylidenequinate (15)Sodium methoxide (28% solution in MeOH, 3.08 mL, 16.0 mmol) was added dropwise to a solution of 14 (4.47 g, 17.6 mmol) in MeOH (94 mL) at 0 °C, and then the mixture was stirred at room temperature for 5 h. The reaction mixture was neutralized with Dowex 50WX8 (H+), and then the resin was separated. The filtrate was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 2 : 1–1 : 2) to give 15 as a yellow oil (4.75 g, 95%). 1H-NMR (CDCl3) δ: 4.46–4.42 (1H, m), 4.13–4.07 (1H, m), 3.96 (1H, t, J = 6.2 Hz), 3.78 (3H, s), 3.49 (1H, s), 2.37 (1H, d, J = 3.2 Hz), 2.29–2.19 (2H, m), 2.07 (1H, ddd, J = 13.7 Hz, 4.2 Hz, 1.4 Hz), 1.84 (1H, dd, J = 13.7 Hz, 10.6 Hz), 1.75–1.51 (8H, m), 1.43–1.33 (2H, m); 13C-NMR (CDCl3) δ: 175.3, 109.9, 79.4, 74.0, 73.0, 68.5, 53.0, 38.9, 37.9, 34.8, 34.6, 24.9, 23.9, 23.5; HR-MS (ESI) m/z: 287.1463 [M + H]+ (Calcd for C14H23O6: 287.1495).
Methyl 4,5-O-Cyclohexylidene-3-dehydro-4-epi-shikimate (16)Dess–Martin periodinane (6.73 g, 15.9 mmol) was added to a solution of 15 (4.13 g, 14.4 mmol) in CH2Cl2 (74 mL) at 0 °C, and then the mixture was stirred at room temperature for 40 h. After saturated NaHCO3 aqueous solution (60 mL) was added the reaction mixture at 0 °C, the mixture was partitioned between CHCl3 and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. After the residue was dissolved in pyridine (15 mL), phosphoryl chloride (2.8 mL, 29.8 mmol) was added to the solution at 0 °C. The resulting mixture was stirred at room temperature for 3 h. After saturated NH4Cl aqueous solution was added the reaction mixture, the mixture was partitioned between CH2Cl2 and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 15 : 1–5 : 1) to give 16 as a pale yellow solid (3.59 g, 97%). 1H-NMR (CDCl3) δ: 6.84 (1H, d, J = 2.7 Hz), 4.69 (1H, dt, J = 5.0, 1.7 Hz), 4.30 (1H, d, J = 5.0 Hz), 3.85 (3H, s), 3.24 (1H, d, J = 20.3 Hz), 2.86 (1H, ddd, J = 20.3, 5.0, 2.7 Hz), 1.69–1.35 (10H, m); 13C-NMR (CDCl3) δ: 197.7, 166.2, 144.3, 131.2, 110.2, 74.7, 72.1, 52.8, 37.0, 35.2, 27.0, 26.6, 24.8, 23.7; HR-MS (ESI) m/z: 267.1214 [M + H]+ (Calcd for C14H19O5: 267.1232).
(1R,2R,3S)-1,2-O-Cyclohexylidene-5-hydroxymethyl-4-cyclohexene-1,2,3-triol (17)Diisobutylaluminium hydride (1.0 M solution in n-hexane, 81.0 mL, 81.0 mmol) was added dropwise to a solution of 16 (3.59 g, 13.5 mmol) in THF (79 mL) at 0 °C, and then the mixture was stirred at room temperature for 3 h. After saturated NH4Cl aqueous solution was added the reaction mixture, the resulting precipitate was filtered off and the filtrate was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc/MeOH, 1 : 4 : 0–0 : 2 : 1) to give 17 as a colorless solid (2.90 g, 90%). 1H-NMR (DMSO-d6) δ: 5.50 (1H, d, J = 0.8 Hz), 4.82 (1H, d, J = 6.0 Hz), 4.70 (1H, t, J = 5.2 Hz), 4.45–4.41 (1H, m), 4.34–4.30 (1H, m), 3.96–3.92 (1H, m), 3.78 (2H, d, J = 5.2 Hz), 2.02 (1H, dd, J = 16.0, 2.0 Hz), 1.84–1.78 (1H, m), 1.51–1.22 (10H, m); 13C-NMR (CDCl3) δ: 137.3, 125.6, 109.4, 76.0, 72.1, 67.0, 65.7, 35.6, 34.0, 28.7, 25.1, 23.9, 23.5; HR-MS (ESI) m/z: 263.1262 [M + Na]+ (Calcd for C13H20NaO4: 263.1259).
(1R,2R,3S)-1,2-O-Cyclohexylidene-3-O-(4-methoxybenzyl)-5-(4-methoxybenzyl-oxy)methyl-4-cyclohexene-1,2,3-triol (18)A solution of 17 (1.20 g, 5.0 mmol) in N,N-dimethylformamide (DMF) (10 mL) was added to a suspention of sodium hydride (60% dispersion in paraffin oil, 516 mg, 15.0 mmol) in DMF (10 mL) at 0 °C, and then the mixture was stirred at room temperature for 1 h. p-Methoxybenzyl chloride (1.67 mL, 15.0 mmol) and n-tetrabutylammonium iodide (369 mg, 1.0 mmol) were added to the reaction mixture at 0 °C, and then the mixture was stirred at room temperature for 20 h. After MeOH was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 4 : 1) to give 18 as a pale yellow oil (2.28 g, 95%). 1H-NMR (CDCl3) δ: 7.32 (2H, d, J = 8.4 Hz), 7.27 (2H, d, J = 8.8 Hz), 6.88 (2H, d, J = 8.4 Hz), 6.87 (2H, d, J = 8.8 Hz), 5.87 (1H, s), 4.73 (1H, d, J = 11.8 Hz), 4.62 (1H, d, J = 11.8 Hz), 4.56 (1H, ddd, J = 7.2 Hz, 3.7 Hz, 1.3 Hz), 4.51–4.48 (1H, m), 4.48 (1H, d, J = 11.2 Hz), 4.37 (1H, d, J = 11.2 Hz), 3.95 (2H, dd, J = 24.4 Hz, 12.4 Hz), 3.82–3.78 (7H, m), 2.41 (1H, dd, J = 16.0 Hz, 1.6 Hz), 1.88–1.82 (1H, m), 1.66–1.41 (8H, m), 1.33–1.24 (2H, m); 13C-NMR (CDCl3) δ: 159.2, 159.1, 134.9, 130.4, 130.1, 129.5, 129.4, 125.0, 113.7, 113.7, 109.2, 74.9, 73.4, 72.9, 72.6, 71.4, 70.4, 55.2(2), 35.6, 33.7, 29.4, 25.3, 23.9, 23.6; HR-MS (ESI) m/z: 503.2427 [M + Na]+ (Calcd for C29H36NaO6: 503.2410).
(1R,2R,3S,4R,5R)-1,2-O-Cyclohexylidene-3-O-(4-methoxybenzyl)-5-(4-methoxybenzyloxymethyl)cyclohexane-1,2,3,4-tetraol (19)Borane-THF complex (1.0 M solution in THF, 13.3 mL, 13.3 mmol) was added to a solution of 18 (3.20 g, 6.66 mmol) in THF (71 mL) at 0 °C, and then the mixture was stirred at room temperature for 20 h. H2O (8.04 mL), H2O2 (35% solution in H2O, 21.8 mL) and 1 M NaOH (51 mL) were added to the reaction mixture at 0 °C, and then the mixture was stirred at room temperature for 3 h. After H2O was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 3 : 1–1 : 1) to give 19 as a colorless oil (2.71 g, 82%). 1H-NMR (CDCl3) δ: 7.32 (2H, d, J = 8.6 Hz), 7.24 (2H, d, J = 8.8 Hz), 6.88 (2H, d, J = 8.6 Hz), 6.86 (2H, d, J = 8.8 Hz), 4.73 (1H, d, J = 12.0 Hz), 4.64 (1H, d, J = 12.0 Hz), 4.44 (2H, s), 4.31 (1H, dd, J = 4.8 Hz, 4.3 Hz), 4.15–4.08 (1H, m), 3.82–3.74 (7H, m), 3.61 (1H, dd, J = 9.2 Hz, 5.6 Hz), 3.45 (1H, dd, J = 9.2 Hz, 6.4 Hz), 3.40 (1H, dd, J = 9.4 Hz, 4.3 Hz), 2.94 (1H, s), 1.90 (1H, ddd, J = 13.8 Hz, 6.2 Hz, 3.6 Hz), 1.77–1.36 (12H, m); 13C-NMR (CDCl3) δ: 159.3, 159.1, 130.2, 129.6, 129.2, 113.8, 113.7, 109.9, 80.0, 77.2, 73.6, 73.4, 72.9, 72.0, 71.4, 70.9, 55.2(2), 38.4, 38.0, 35.1, 30.7, 25.0, 24.0, 23.7; HR-MS (ESI) m/z: 521.2534 [M + Na]+ (Calcd for C29H38NaO7: 521.2515).
(1R,2R,3S,4R,5R)-4-O-Acetyl-1,2-O-cyclohexylidene-3-O-(4-methoxybenzyl)-5-(4-methoxybenzyloxymethyl)cyclohexane-1,2,3,4-tetraol (20)Acetic anhydride (6.48 mL, 68.5 mmol) was added to a solution of 19 (1.71 g, 3.43 mmol) in pyridine (15 mL) at 0 °C, and then the mixture was stirred at room temperature for 20 h. After saturated NaHCO3 aqueous solution was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 3 : 1) to give 20 as a colorless oil (1.80 g, 97%). 1H-NMR (CDCl3) δ: 7.26 (2H, d, J = 8.6 Hz), 7.22 (2H, d, J = 8.6 Hz), 6.86 (4H, d×2, J = 8.6 Hz), 5.16 (1H, dd, J = 10.4 Hz, 9.2 Hz), 4.36 (2H, dd, J = 16.0 Hz, 11.6 Hz), 4.28 (1H, dd, J = 4.8 Hz, 4.4 Hz), 4.15–4.09 (1H, m), 3.80 (6H, s×2), 3.55 (1H, dd, J = 9.2 Hz, 4.4 Hz), 3.41 (1H, dd, J = 9.2 Hz, 4.8 Hz), 3.27 (1H, dd, J = 9.2 Hz, 7.2 Hz), 2.05–1.98 (4H, m), 1.80–1.50 (10H, m), 1.41–1.36 (2H, m); 13C-NMR (CDCl3) δ: 170.3, 159.2, 159.1, 130.4, 130.2, 129.3, 129.2, 113.7, 113.6, 110.0, 77.2, 77.1, 74.2, 73.1, 72.9, 72.0, 71.5, 71.3, 55.2(2), 37.7, 35.0, 30.5, 23.9, 25.0, 23.9, 21.1; HR-MS (ESI) m/z: 563.2636 [M + Na]+ (Calcd for C31H40NaO8: 563.2621).
(1R,2R,3S,4R,5R)-4-O-Acetyl-3-O-(4-methoxybenzyl)-5-(4-methoxybenzyloxy-methyl)cyclohexane-1,2,3,4-tetraol (21)At 0 °C, 20 (1.65 g, 3.05 mmol) was suspended in a solution of acetic acid (8.4 mL) and H2O (2.1 mL). The resulting mixture was stirred at room temperature for 17 h. The reaction mixture was concentrated under reduced pressure. The residue was partitioned between CH2Cl2 and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 2 : 3) to give 21 as a colorless solid (1.31 g, 94%). 1H-NMR (CDCl3) δ: 7.22 (2H, d, J = 8.6 Hz), 7.20 (2H, d, J = 8.8 Hz), 6.87 (2H, d, J = 8.6 Hz), 6.85 (2H, d, J = 8.8 Hz), 5.11 (1H, t, J = 10.0 Hz), 4.56 (1H, d, J = 12.0 Hz), 4.49 (1H, d, J = 12.0 Hz), 4.36 (2H, dd, J = 20.6 Hz, 11.4 Hz), 4.14–4.11 (1H, m), 3.81 (3H, s), 3.80 (3H, s), 3.64–3.57 (1H, m), 3.38 (1H, dd, J = 9.3 Hz, 4.0 Hz), 3.34 (1H, dd, J = 9.6 Hz, 2.8 Hz), 3.26 (1H, dd, J = 9.3 Hz, 6.2 Hz), 2.52 (1H, s), 2.20 (1H, d, J = 10.4 Hz), 1.97–1.94 (4H, m), 1.79–1.64 (2H, m); 13C-NMR (CDCl3) δ: 170.5, 159.3 159.1, 130.2, 129.7, 129.3, 129.3, 113.8, 113.7, 79.7, 72.9, 71.9, 71.6, 70.8, 70.2, 69.3, 55.2(2), 37.7, 30.6, 21.1; HR-MS (ESI) m/z: 460.1763 [M + K]+ (Calcd for C25H32KO8: 460.1734).
(1R,2S,3S,4R,5R)-4-O-Acetyl-2-O-benzoyl-3-O-(4-methoxybenzyl)-5-(4-methoxy-benzyloxymethyl)cyclohexane-1,2,3,4-tetraol (22)Benzoic acid (708 mg, 5.77 mmol), triphenylphosphine (1.50 g, 5.77 mmol) and MS4A (220 mg) were added to a suspension of 21 (442 mg, 962 µmol) in toluene (19 mL) at 0 °C. Diisopropyl azodicarboxylate (1.10 mL, 5.77 mmol) was added dropwise to the mixture at 0 °C, and then the resuting mixture was refluxed for 18 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 3 : 1–2 : 1) to give 22 as a colorless solid (308 mg, 57%). 1H-NMR (CDCl3) δ: 8.03 (2H, d, J = 7.6 Hz), 7.58 (1H, t, J = 7.6 Hz), 7.44 (2H, t, J = 7.6 Hz), 7.24 (2H, d, J = 8.4 Hz), 7.00 (2H, d, J = 8.6 Hz), 6.87 (2H, d, J = 8.4 Hz), 6.67 (2H, d, J = 8.6 Hz), 5.16 (1H, t, J = 9.4 Hz), 5.06 (1H, dd, J = 10.8 Hz, 9.6 Hz), 4.53 (2H, dd, J = 14.0 Hz, 10.8 Hz), 4.38 (2H, dd, J = 16.8 Hz, 11.2 Hz), 3.81 (3H, s), 3.83–3.75 (1H, m), 3.71–3.65 (4H, m), 3.43 (1H, dd, J = 9.2 Hz, 4.4 Hz), 3.30 (1H, dd, J = 9.2 Hz, 6.4 Hz), 2.30 (1H, d, J = 5.2 Hz), 2.26 (1H, dt, J = 13.0 Hz, 4.2 Hz), 1.98–1.88 (4H, m), 1.48 (1H, q, J = 13.0 Hz); 13C-NMR (CDCl3) δ: 170.6, 166.8, 159.2, 159.0, 133.2, 130.1, 130.0, 129.8, 129.6, 129.4, 129.3, 128.4, 113.7, 113.6, 80.8, 79.3, 74.2, 74.1, 73.0, 70.8, 70.4, 55.3, 55.1, 37.8, 33.1, 20.9; HR-MS (ESI) m/z: 587.2232 [M + Na]+ (Calcd for C32H36NaO9: 587.2257).
(2R,3S,4R,5R)-4-O-Acetyl-2-O-benzoyl-3-O-(4-methoxybenzyl)-5-(4-methoxy-benzyloxymethyl)cyclohexane-2,3,4-triol-1-on (9)Dess–Martin periodinane (275 mg, 648 µmol) was added to a solution of 22 (182 mg, 324 µmol) in CH2Cl2 (4.5 mL), and then the mixture was stirred at room temperature for 2 h. After saturated NaHCO3 aqueous solution was added the reaction mixture at 0 °C, the mixture was partitioned between CHCl3 and H2O. The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 2 : 1) to give 9 as a colorless solid (180 mg, 99%). 1H-NMR (CDCl3) δ: 8.04 (2H, d, J = 7.6 Hz), 7.59 (1H, t, J = 7.6 Hz), 7.46 (2H, t, J = 7.6 Hz), 7.24 (2H, d, J = 8.8 Hz), 7.12 (2H, d, J = 8.6 Hz), 6.88 (2H, d, J = 8.8 Hz), 6.77 (2H, d, J = 8.6 Hz), 5.57 (1H, d, J = 10.8 Hz), 5.51 (1H, dd, J = 11.2 Hz, 9.6 Hz), 4.70 (1H, d, J = 11.0 Hz), 4.58 (1H, d, J = 11.0 Hz), 4.39 (2H, dd, J = 16.4 Hz, 11.6 Hz), 3.91 (1H, t, J = 9.8 Hz), 3.81 (3H, s), 3.75 (3H, s), 3.40 (2H, d, J = 4.4 Hz), 2.65 (1H, t, J = 14.0 Hz), 2.61 (1H, dd, J = 14.0 Hz, 5.2 Hz), 2.12–2.02 (1H, m), 1.96 (3H, s); 13C-NMR (CDCl3) δ: 199.6, 169.8, 165.1, 159.3(2), 133.3, 129.9, 129.8, 129.7, 129.5(2), 129.2, 128.4, 113.7(2), 80.8, 79.6, 74.4, 73.0, 72.4, 68.7, 55.2, 55.2, 39.4, 37.9, 20.8; HR-MS (ESI) m/z: 587.2090 [M + Na]+ (Calcd for C32H34NaO9: 585.2101).
(1R,2R,3S,4R,5R)-4-O-Acetyl-2-O-benzoyl-1-ethynyl-3-O-(4-methoxybenzyl)-5-(4-methoxy-benzyloxymethyl)cyclohexane-1,2,3,4-tetraol (23) and (1S,2R,3S,4R,5R)-4-O-Acetyl-2-O-benzoyl-1-ethynyl-3-O-(4-methoxybenzyl)-5-(4-methoxy-benzyloxymethyl)cyclohexane-1,2,3,4-tetraol (24)Ethynylmagnesium bromide (0.5 M solution in THF, 6.08 mL, 3.04 mmol) was added to a solution of 9 (532 mg, 950 µmol) in THF (6 mL) at −80 °C, and then the mixture was stirred at −40 °C for 24 h. After saturated NH4Cl aqueous solution was added to the reaction mixture at −40 °C, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 4 : 1) to give a 1 : 5.6 mixtute of 23 and 24 (542 mg, 97%) as a pale yellow solid. The mixture was then separated by column chromatography on silica gel (CHCl3/n-hexane, 50 : 1).
23: 1H-NMR (CDCl3) δ: 8.03 (2H, d, J = 7.4 Hz), 7.59 (1H, t, J = 7.4 Hz), 7.46 (2H, t, J = 7.4 Hz), 7.26 (2H, d, J = 8.6 Hz), 6.96 (2H, d, J = 8.4 Hz), 6.88 (2H, d, J = 8.6 Hz), 6.63 (2H, d, J = 8.4 Hz), 5.44 (1H, d, J = 10.0 Hz), 5.14 (1H, t, J = 10.0 Hz), 4.46 (2H, dd, J = 18.0 Hz, 11.2 Hz), 4.37 (2H, dd, J = 17.4 Hz, 11.2 Hz), 3.95 (1H, t, J = 10.0 Hz), 3.81 (3H, s), 3.69 (3H, s), 3.39 (1H, dd, J = 9.3 Hz, 3.8 Hz), 3.32 (1H, dd, J = 9.3 Hz, 5.6 Hz), 2.60 (1H, s), 2.36 (1H, s), 2.36–2.27 (2H, m), 1.93 (3H, s), 1.86 (1H, t, J = 14.0 Hz); 13C-NMR (CDCl3) δ: 170.0, 164.9, 159.1, 158.9, 133.1, 130.1, 130.1, 129.7, 129.6, 129.4, 129.2, 128.3, 113.6, 113.5, 83.6, 79.1, 77.4, 74.3, 73.8, 73.0, 72.9, 69.6, 68.7, 55.2, 55.0, 37.2, 36.0, 20.9; HR-MS (ESI) m/z: 611.2247 [M + Na]+ (Calcd for C34H36NaO9: 611.2257).
24: 1H-NMR (CDCl3) δ: 8.08 (2H, d, J = 7.4 Hz), 7.59 (1H, t, J = 7.4 Hz), 7.46 (2H, t, J = 7.4 Hz), 7.25 (2H, d, J = 8.2 Hz), 6.99 (2H, d, J = 8.6 Hz), 6.88 (2H, d, J = 8.2 Hz), 6.66 (2H, d, J = 8.6 Hz), 5.21 (1H, d, J = 9.6 Hz), 5.11 (1H, dd, J = 11.2 Hz, 9.6 Hz), 4.51 (2H, dd, J = 17.0 Hz, 11.0 Hz), 4.38 (2H, dd, J = 15.4 Hz, 11.0 Hz), 3.90 (1H, t, J = 9.6 Hz), 3.81 (3H, s), 3.71 (3H, s), 3.43 (1H, dd, J = 9.2 Hz, 4.0 Hz), 3.33 (1H, dd, J = 9.2 Hz, 5.6 Hz), 2.97 (1H, s), 2.68 (1H, s), 2.30–2.21 (2H, m), 1.92 (3H, s), 1.78 (1H, t, J = 14.0 Hz); 13C-NMR (CDCl3) δ: 170.0, 166.4, 159.1, 159.0, 133.4, 130.1, 130.0, 129.9, 129.5, 129.4, 129.2, 128.4, 113.7, 113.6, 82.7, 80,6, 79.0, 75.5, 74.5, 73.8, 73.0, 70.7, 69.9, 55.2, 55.1, 38.3, 37.6, 20.9; HR-MS (ESI) m/z: 611.2265 [M + Na]+ (Calcd for C34H36NaO9: 611.2257).
(1R,2R,3S,4R,5R)-1-Ethynyl-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (1-Ethynyl-α-D-carbaglucose) (10)H2O (322 µL) and 2,3-dichloro-5,6-dicyano-p-benzoquinone (119 mg, 525 µmol) were added to a solution of 23 (155 mg, 262 µmol) in CH2Cl2 (5.3 mL), and then the mixture was stirred at room temperature for 24 h. The reaction mixture was dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/acetone, 3 : 1 then CHCl3/methanol, 30 : 1) to give the corresponding triol. Sodium methoxide (25.2 mg, 460 µmol) was added to a solution of the triol in MeOH (5.5 mL), and then the mixture was stirred at room temperature for 2.5 h. The reaction mixture was neutralized with Dowex 50WX8 (H+), and then the resin was separated. The filtrate was washed with EtOAc. The water layer was concentrated under reduced pressure to give 10 as a colorless solid (43.5 mg, 81%). 1H-NMR (CD3OD) δ: 3.62–3.60 (2H, m), 3.46–3.41 (1H, m), 3.28–3.19 (2H, m), 2.74 (1H, s), 2.03 (1H, dd, J = 13.9 Hz, 3.6 Hz), 1.86–1.79 (1H, m), 1.54 (1H, t, J = 13.9 Hz); 13C-NMR (CD3OD) δ: 87.6, 78.7, 76.5, 74.6, 72.5, 70.4, 63.7, 40.1, 38.9; HR-MS (ESI) m/z: 225.0721 [M + Na]+ (Calcd for C9H14NaO5: 225.0739).
(1S,2R,3S,4R,5R)-1-Ethynyl-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (1-Ethynyl-β-D-carbaglucose) (11)H2O (534 µL) and 2,3-dichloro-5,6-dicyano-p-benzoquinone (247 mg, 1.09 µmol) were added to a solution of 24 (256 mg, 435 µmol) in CH2Cl2 (8.8 mL), and then the mixture was stirred at room temperature for 23 h. The reaction mixture was dried over Na2SO4, and then concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/acetone, 3 : 1 then CHCl3/methanol, 30 : 1) to give the corresponding triol. Sodium methoxide (50.7 mg, 938 µmol) was added to a solution of the triol in MeOH (5.2 mL), and then the mixture was stirred at room temperature for 2.5 h. The reaction mixture was neutralized with Dowex 50WX8 (H+), and then the resin was separated. The filtrate was washed with EtOAc. The water layer was concentrated under reduced pressure to give 11 as a colorless solid (94.9 mg, quant). 1H-NMR (CD3OD) δ: 3.72 (1H, dd, J = 10.8 Hz, 3.6 Hz), 3.61 (1H, dd, J = 10.8 Hz, 6.0 Hz), 3.27–3.18 (2H, m), 2.92 (1H, s), 2.02 (1H, dd, J = 13.3 Hz, 3.8 Hz), 1.88–1.82 (1H, m), 1.44 (1H, t, J = 13.3 Hz); 13C-NMR (CD3OD) δ: 85.1, 79.8, 77.8, 76.3, 74.6, 72.2, 63.8, 42.0, 39.1; HR-MS (ESI) m/z: 225.0725 [M + Na]+ (Calcd for C9H14NaO5: 225.0739).
4-Ethylbenzylazide (12b)Et3N (1.01 mL, 7.26 mmol) and methanesulfonyl chloride (560 µL, 7.26 mmol) were added to a solution of 4-ethylbenzyl alcohol24) (761 mg, 5.59 mmol) prepared from 4-ethylbenzaldehyde in DMF (4.5 mL) at 0 °C, and the mixture was stirred at room temperature for 18 h. Et3N (1.01 mL, 7.26 mmol) and methanesulfonyl chloride (560 µL, 7.26 mmol) were added to the reaction mixture at 0 °C, and the resulting mixture was stirred at room temperature for 24 h. After saturated NaHCO3 aqueous solution was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O, saturated NaHCO3 aqueous solution and brine, dried over Na2SO4, and concentrated under reduced pressure. After the residue was dissolved in DMF (8.6 mL), sodium azide (1.11 g, 17.1 mmol) was added to the solution. The resulting mixture was stirred at room temperature for 15 h. After saturated NaHCO3 aqueous solution was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane/EtOAc, 10 : 1) to give 12b as a colorless oil (585 mg, 65%). 1H-NMR (CDCl3) δ: 7.30–7.26 (4H, m), 4.35 (2H, s), 2.71 (2H, q, J = 6.1 Hz), 1.30 (3H, t, J = 6.1 Hz); 13C-NMR (CDCl3) δ: 144.4, 132.5, 128.3(2), 54.6, 28.5, 15.5; HR-MS (ESI) m/z: 162.1055 [M + H]+ (Calcd for C9H12N3: 162.1031).
4-Cyclopropylbenzylazide (12c)Et3N (767 µL, 5.50 mmol) and methanesulfonyl chloride (424 µL, 5.50 mmol) were added to a solution of 4-cyclopropylbenzyl alcohol25) (627 mg, 4.23 mmol) prepared from 4-bromobenzyl alcohol in DMF (3.4 mL) at 0 °C, and the mixture was stirred at room temperature for 18 h. After saturated NaHCO3 aqueous solution was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O, saturated NaHCO3 aqueous solution and brine, dried over Na2SO4, and concentrated under reduced pressure. After the residue was dissolved in DMF (7.3 mL), sodium azide (1.29 g, 19.9 mmol) was added to the solution. The resulting mixture was stirred at room temperature for 18 h. After saturated NaHCO3 aqueous solution was added the reaction mixture, the mixture was partitioned between EtOAc and H2O. The organic layer was washed with H2O and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (n-hexane) to give 12c as a colorless oil (568 mg, 77%). 1H-NMR (CDCl3) δ: 7.20 (2H, d, J = 8.2 Hz), 7.08 (2H, d, J = 8.2 Hz), 4.28 (2H, s), 1.94–1.87 (1H, m), 1.00–0.95 (2H, m), 0.72–0.6 (2H, m); 13C-NMR (CDCl3) δ: 143.4, 135.3, 127.9, 125.6, 71.7, 15.2, 9.2; HR-MS (ESI) m/z: 174.1062 [M + H]+ (Calcd for C10H12N3: 174.1031).
General Procedure for CuAAC Reaction between 1-Ethynyl-carbaglucose and Azide for Synthesis of 7 and 8CuSO4 (40 mM in H2O, 249 µL, 10 µmol) and sodium ascorbate (100 mM in H2O, 100 µL, 10 µmol) were added to a solution of α- or β-1-ethynyl-carbaglucose (20.2 mg, 100 µmol) and azide 12 (500 µmol) in DMF (370 µL), and then the mixture was stirred at room temperature. The reaction mixture was concentrated under reduced pressure, the resulting residue was purified by column chromatography on silica gel (CHCl3/MeOH, 30 : 1–1 : 1) to give 7 or 8.
(1R,2R,3S,4R,5R)-1-[1-Benzyl-1H-1,2,3-triazol-4-yl]-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (7a)90%; 1H-NMR (CD3OD) δ: 7.85 (1H, s), 7.37–7.30 (5H, m), 5.55 (2H, s), 3.73–3.56 (3H, m), 3.39–3.29 (2H, m), 2.07–1.81 (3H, m); 13C-NMR (CD3OD) δ: 155.3, 136.8, 130.0, 129.6, 129.2, 123.9, 78.1, 77.2, 74.9, 73.8, 64.2, 54.9, 40.5, 38.1; HR-MS (ESI) m/z: 358.1367 [M + Na]+ (Calcd for C16H21NaN3O5: 358.1379).
(1R,2R,3S,4R,5R)-1-[1-(4-Ethylbenzyl)-1H-1,2,3-triazol-4-yl]-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (7b)quant; 1H-NMR (CD3OD) δ: 7.80 (1H, s), 7.26 (2H, d, J = 8.2 Hz), 7.20 (2H, d, J = 8.2 Hz), 5.51 (2H, s), 3.78–3.57 (3H, m), 3.38–3.26 (2H, m), 2.62 (2H, q, J = 7.6 Hz), 2.00–1.81 (2H, m), 1.19 (3H, t, J = 7.6 Hz); 13C-NMR (CD3OD) δ: 155.2, 146.1, 134.0, 129.5, 129.4, 123.7, 78.0, 77.2, 74.9, 73.7, 64.1, 54.8, 40.5, 38.1, 29.5, 16.1; HR-MS (ESI) m/z: 386.1684 [M + Na]+ (Calcd for C18H25NaN3O5: 386.1692).
(1R,2R,3S,4R,5R)-1-[1-(4-Cyclopropylbenzyl)-1H-1,2,3-triazol-4-yl]-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (7c)quant; 1H-NMR (CD3OD) δ: 7.77 (1H, s), 7.22 (2H, d, J = 8.4 Hz), 7.06 (2H, d, J = 8.4 Hz), 5.49 (2H, s), 3.73–3.56 (4H, m), 3.38–3.33 (1H, m), 2.05–1.80 (4H, m), 0.97–0.93 (2H, m), 0.66–0.63 (2H, m); 13C-NMR (CD3OD) δ: 155.2, 146.1, 133.6, 129.4, 127.1, 123.6, 78.1, 77.2, 75.0, 73.7, 64.2, 54.7, 40.5, 38.1, 15.9, 9.8; HR-MS (ESI) m/z: 398.1675 [M + Na]+ (Calcd for C19H25NaN3O5: 398.1692).
(1S,2R,3S,4R,5R)-1-[1-Benzyl-1H-1,2,3-triazol-4-yl]-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (8a)quant; 1H-NMR (CD3OD) δ: 8.07 (1H, s), 7.38–7.30 (5H, m), 5.58 (2H, s), 3.71–3.61 (2H, m), 3.54–3.47 (1H, m), 3.36–3.25 (2H, m), 2.56–2.52 (1H, m), 1.75–1.70 (1H, m), 1.64–1.58 (1H, m); 13C-NMR (CD3OD) δ: 136.9, 130.0, 129.5, 129.2, 125.8, 80.8, 77.4, 75.2, 73.3, 71.5, 64.0, 54.8, 41.5, 37.7; HR-MS (ESI) m/z: 358.1366 [M + Na]+ (Calcd for C16H21NaN3O5: 358.1379).
(1S,2R,3S,4R,5R)-1-[1-(4-Ethylbenzyl)-1H-1,2,3-triazol-4-yl]-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (8b)94%; 1H-NMR (CD3OD) δ: 8.03 (1H, s), 7.25 (2H, d, J = 8.2 Hz), 7.19 (2H, d, J = 8.2 Hz), 5.52 (2H, s), 3.70–3.61 (2H, m), 3.50 (1H, d, J = 9.6 Hz), 3.36–3.24 (2H, m), 2.62 (2H, q, J = 7.6 Hz), 2.54 (1H, dd, J = 13.2 Hz, 3.4 Hz), 1.76–1.68 (1H, m), 1.60 (1H, t, J = 13.2 Hz), 1.20 (3H, t, J = 7.6 Hz); 13C-NMR (CD3OD) δ: 146.0, 134.1, 129.5, 129.3, 125.7, 80.7, 77.3, 75.2, 73.3, 71.3, 64.0, 54.6, 41.5, 37.7, 29.5, 16.1; HR-MS (ESI) m/z: 386.1673 [M + Na]+ (Calcd for C18H25NaN3O5: 386.1692).
(1S,2R,3S,4R,5R)-1-[1-(4-Cyclopropylbenzyl)-1H-1,2,3-triazol-4-yl]-5-hydoxymethylcyclohexane-1,2,3,4-tetraol (8c)88%; 1H-NMR (CD3OD) δ: 8.02 (1H, s), 7.21 (2H, d, J = 5.0 Hz), 7.06 (2H, d, J = 5.0 Hz), 5.50 (2H, s), 3.70–3.47 (3H, m), 3.35–3.23 (2H, m), 2.56–2.52 (1H, m), 1.92–1.85 (1H, m), 1.76–1.67 (1H, m), 1.63–1.57 (1H, m), 0.97–0.93 (2H, m), 0.67–0.63 (2H, m); 13C-NMR (CD3OD) δ: 146.0, 133.6, 129.2, 127.0, 125.7, 80.7, 77.3, 75.2, 73.3, 71.3, 64.0, 54.6, 41.5, 37.7, 15.9, 9.9; HR-MS (ESI) m/z: 398.1668 [M + Na]+ (Calcd for C19H25NaN3O5: 398.1692).
X-ray Crystallographic Structure Determination for 24X-ray diffraction data for 24 was collected using a Rigaku Saturn 724 CCD diffractometer with Mo-Kα radiation (l = 0.71073 Å) at 123 K. Single crystals (size: 0.10 × 0.10 × 0.05 mm3) of 24 (C34H36O9, Mw = 588.63) suitable for X-ray analysis were grown by the recrystallization of a solution of 24 in CH2Cl2/n-hexane (1 : 5) at –20 °C for approximately 2 weeks. The unit cell was monoclinic with the space group P21. Lattice constants with Z = 4, ρcalcd = 1.307 g/cm3, μ = 0.094 cm−1, F(000) = 1248, θmax = 27.500° were a = 14.7668(8), b = 10.4299(6), c = 19.5056(11) Å, α = 90°, β = 95.101(5)°, γ = 90°, and V = 2992.3(3) Å3. A total of 41,986 reflections were collected, of which 13,713 reflections were independent (Rint = 0.0831). The structure was refined to final R1 = 0.0640 for 8,218 data [I > 2σ(I)] with 787 parameters and wR2 = 0.1409 for all data, GOF = 1.000, and residual electron density max./min. = 0.282/−0.222 e·Å−3. The ORTEP diagram is shown in Supplementary Fig. S1, and the crystal data and structure refinement are listed in Supplementary Table S1. Data collection, cell refinement, and data reduction were conducted using the CrystalClear-SM Expert 2.0 software.26) The structure was solved by direct methods using the program SHELXT27) and refined by full-matrix least-squares methods on F2 using SHELXL2014.28) All materials for publication were prepared by Yadokari-XG 2009 software.29) All non-hydrogen atoms were refined anisotropically. Tables of positional and thermal parameters, bond lengths and angles, torsion angles and Figs. may be found from the Cambridge Crystallographic Centre by referencing CCDC number 2182979.
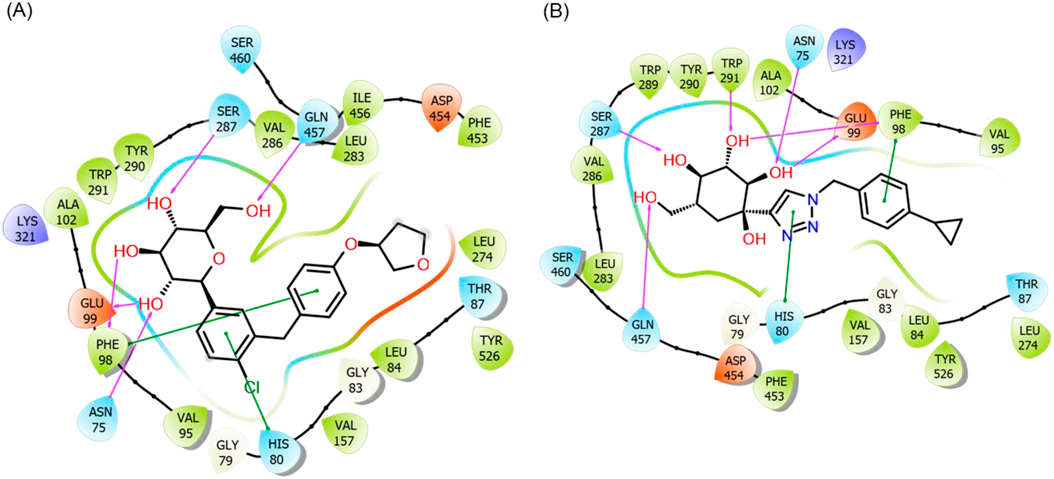
Molecular Docking StudyDocking investigations, protein and chemical preparations were carried out in accordance with earlier descriptions.30,31) The compounds’ 2 dimensional (2D) structures were imported and 3D optimized by LigPrep software using the OPLS2005 force field. The protein preparation wizard in the Maestro suite (Schrodinger LLC, NY, U.S.A.) was used to process and optimize the SGLT2 structure (PDB ID 7vsi). The structure was checked, protonated, optimized at physiological pH and corrected for missing atoms or side chains by using Prime software implemented in the Schrodinger suite. The OPLS2005 force field was used for energy minimization. All docking and molecular dynamics calculations in this investigation were based on the generated structure. The standard precision docking approach was used to dock all compounds (SP docking). In the docking grid, the co-crystallized ligand, empagliflozin (4), functioned as the center of a 20 Å docking box that ringed the bound ligand. Redocking of the co-crystalized ligand, 4 was employed as a quality check for docking accuracy. The finding of accurate docking of 4 at the beginning of docking operations supported the accuracy of docking protocol.
Establishment of hSGLT2 Stable Cell LinehSGLT2 cDNA (pFN21AE5578) was purchased from Kazusa DNA Research Institute. Gene fragments for the creation of full-length hSGLT2 were synthesized on order and In-Fusion technology was used to construct a full-length hSGLT2 gene expression vector. PCR was also performed to add a Flag tag to the C-terminus of hSGLT2, which was finally assembled into the pF5A-CMV-neo vector. The construct was confirmed by sequencing. HEK293 cell line was maintained as an adherent culture in Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% fetal bovine serum (FBS). The cells were transfected with the plasmid pF5A-CMV-neo-SGLT2-Flag using polyethylenimine-based transfection method. One day after transfection, cells were transferred into fresh D-MEM medium containing 800 µg/mL geneticin and the medium was replaced every three days. Two weeks later, ten cell clones were separately expanded into two 10 cm dishes. Cells in dish were harvested during the exponential growth phase and frozen in 10% DMSO for storage. HEK293 cells stably overexpressing hSGLT2 (hSGLT2/HEK293) were cultured at 37 °C in a humidified atmosphere of 5% CO2 in air in D-MEM supplemented with 10% FBS.
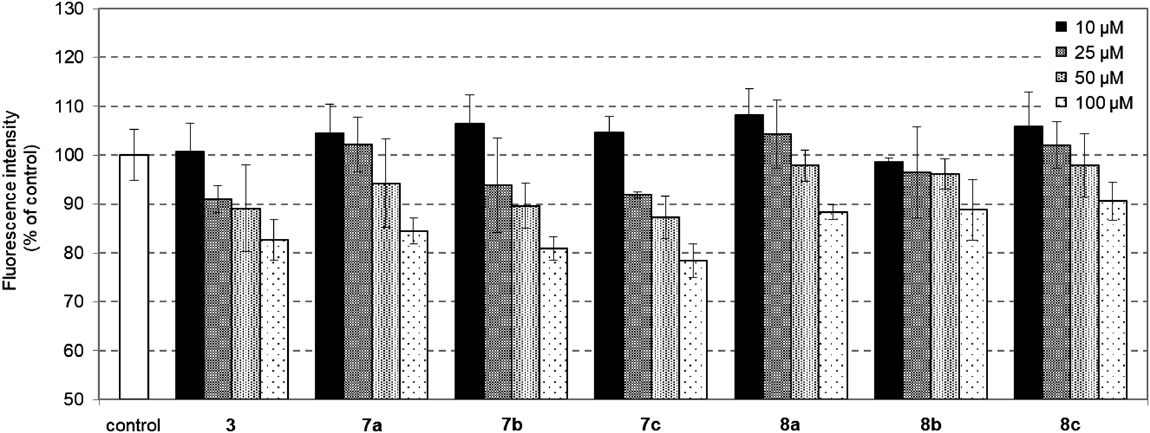
Fluorescent SGLT2 AssayhSGLT2/HEK293 were washed twice with the pretreatment buffer (10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 140 mM choline chloride, 2 mM KCl, 1 mM CaCl2, and 1 mM MgCl2, pH 7.4, pH was adjusted with Tris), then incubated with the uptake buffer (10 mM HEPES, 140 mM NaCl, 2 mM KCl, 1 mM CaCl2, and 1 mM MgCl2, pH 7.4, pH was adjusted with Tris). The cells were incubated with 25 mM 1-azido-1-deoxy-D-glucose (25) in the uptake buffer in the presence of the indicated amount of 3, 7 or 8 in DMSO or DMSO as a control at 37 °C for 2 h. After removing the culture medium, the cells were washed twice with ice-cold washing buffer (the pretreatment buffer containing 100 µM phlorizin) to remove the un-transported 25. Cells were then treated with sodium dodecyl sulfate (SDS) lysis buffer containing CuAAC-activated fluorogenic alkyne probe and CuAAC reagents (100 µM 4-ethynyl-N-ethyl-1,8-naphthalimide, 250 µM tris[(1-ethoxycarbonylmethyl-1H-1,2,3-triazol-4yl)methyl]-amine, 2.5 mM CuSO4·5H2O, 5 mM sodium ascorbate, 0.1% SDS in phosphate buffered saline (PBS)) and incubated at 37 °C for 18 h to afford the desired fluorescence adduct. After a certain amount of each well was centrifuged (15000 rpm × 10 min, 24 °C), the fluorescence intensity of the generated fluorescently labeled triazolyl glycan was measured by a multimode plate reader (TriStar LB941, Berthold Technologies) with an excitation/emission wavelength of 355 nm/460 nm. The percent of inhibition was calculated by comparing fluorescence intensity. The results were confirmed by at least three independent experiments with two cultures each and expressed as an average of mean ± standard deviation (S.D.) from three experiments.
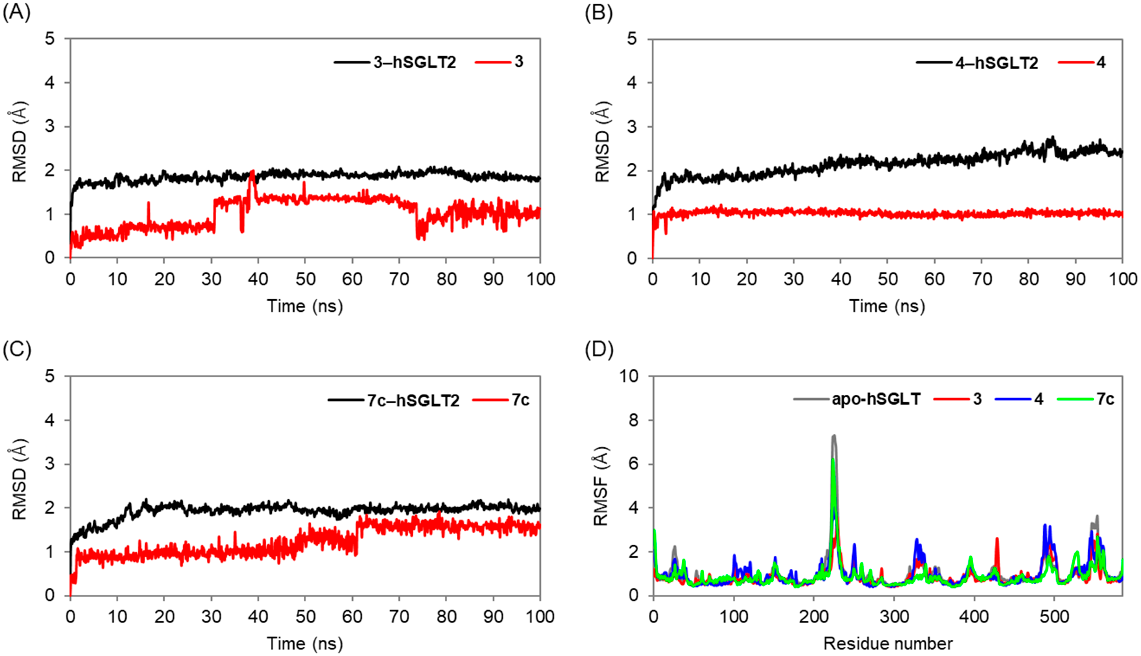
Molecular Dynamics Simulation StudyMolecular dynamics simulations lasting 100 ns were carried out using the Desmond software, Schrödinger LLC (New York, U.S.A.). The system builder tool was used to set up each system. The protein was introduced into a DPPC membrane at 325 K. The hSGLT2 structures embedded in the membrane were solvated in a cubic box of water of the orthorhombic solvent model (transferable intermolecular interaction potential 3 points; TIP3P). The simulation made use of the OPLS 2005 force field. Counter ions were used to neutralize the models. One hundred fifty millimolar NaCl was used to mimic physiological circumstances. One atm pressure and 300 K temperature were maintained for the whole simulation in the isothermal-isobaric ensemble. To minimize the systems, 5000 steepest decline steps were followed by progressive heating from 0 to 300 K. For five ns of temperature relaxation, the Nosé-Hoover Chain thermostat method32–34) and the Martyna-Tobias-Klein barostat method35) for 5 ns of pressure relaxation were used. Coordinates were collected every 100 ps to generate trajectories of 1000 frames. RMSD, RMSF, hydrogen bonding between proteins and bonds distances were studied in the trajectory.