Review
Functions, Structures, and Engineering of the Teleocidin Biosynthetic Enzymes
2023 年 71 巻 3 号 p. 188-197
詳細
2023 年 71 巻 3 号 p. 188-197
Teleocidins are natural products belonging to the indole alkaloid family and show potent protein kinase C activation activity. The structural feature of teleocidins is an indole-fused nine-membered lactam ring structure. Due to their unique structures and strong biological activities, many total synthesis and biosynthetic studies of teleocidins have been performed. Teleocidin biosynthesis involves interesting enzymatic reactions that are challenging in organic synthesis, including oxidative intramolecular C–N bond-forming reactions, regio- and stereo-selective reverse prenylation reactions, and methylation-triggered terpene cyclization. This review summarizes the recent research on functional and structural analyses, as well as enzyme engineering, of teleocidin biosynthetic enzymes.
Teleocidins B1, B2, B3, and B4 (1–4) are unique indole alkaloids, with an indole-fused nine-membered ring (indolactam) and a monoterpenoid moiety fused with C6 and C7 of the indole1–3) (Fig. 1). Teleocidins were first discovered from Streptomyces mediocidicus in 1960, as toxic compounds against fish.4) In 1966, the structures of teleocidin Bs were determined as dihydroteleocidin B bromoacetate derivatives by X-ray crystallography.5) Since then, various teleocidin derivatives have been identified from Streptomyces sp. and the cyanobacterium Moorea producens.6–12) These compounds show 12-O-tetradecanoylphorbol-13-acetate (TPA)-like agonist activities toward classical protein kinase Cs (cPKCs) and novel PKCs (nPKCs).13) Investigations on the structure–activity relationships of teleocidin derivatives revealed that the indolactam V (5) skeleton, the common core structure of teleocidins, is essential for their biological activities.14) Due to their unique structures and activities, many total syntheses, biosynthetic analyses, and derivatizations of teleocidins have been conducted.15–35) This review will summarize the results of studies on the structures and functions of teleocidins, as well as the engineering of enzymes involved in their biosynthesis.
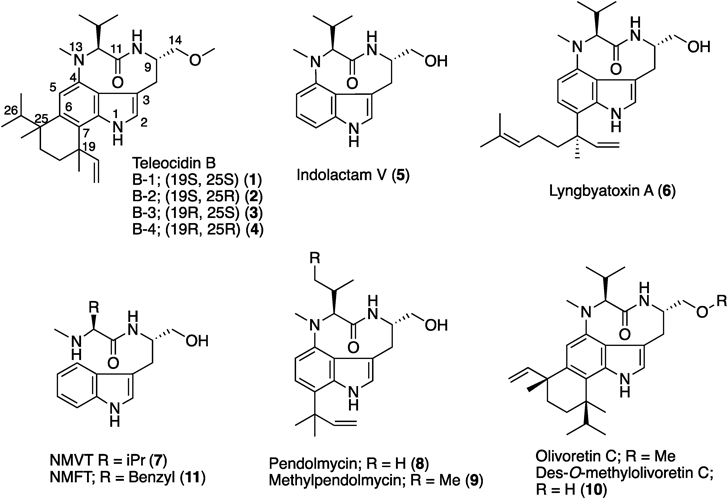
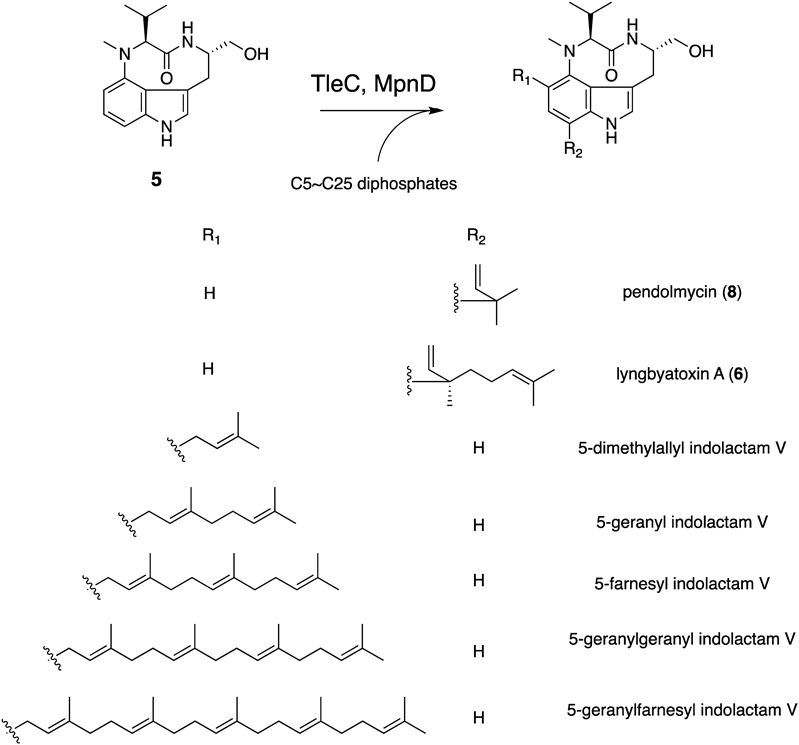
The biosynthetic gene cluster of lyngbyatoxin A (6) (ltxA-D) was identified from the M. producens genome in 2004 by Gerwick and colleagues.30,31) The gene cluster encodes a non-ribosomal peptide synthetase (NRPS, LtxA), a CYP (LtxB), an ABBA-type prenyltransferase (LtxC), and an oxidase/reductase-related protein (LtxD). The domain structure of LtxA is MT-A-T-C-A-T-R (MT: methyltransferase domain, A: adenylation domain, T: thiolation domain, C: condensation domain, and R: reductase domain)32) (Fig. 2). The functional analysis of LtxA revealed that the A domains accept valine and tryptophan as substrates, respectively, and the terminal R domain catalyzes the four electron reduction of a carrier protein-tethered peptide to release N-methyl-L-valyl-L-tryptophanol (NMVT, 7). The in vitro analysis of LtxB indicated that the enzyme catalyzes the C–N bond formation between C4 and N-13 to yield 5. The prenyltransferase LtxC catalyzes the reverse prenylation of a geranyl group to C7 of 5 to generate 6. LtxD was predicted to be involved in the conversion of 6 into the minor metabolites lyngbyatoxins B and C; however, the heterologous expression of the ltx cluster in Escherichia coli and Anabaena sp. strain PCC 7120 did not generate these products.33) Thus, the true function of LtxD remains unknown.
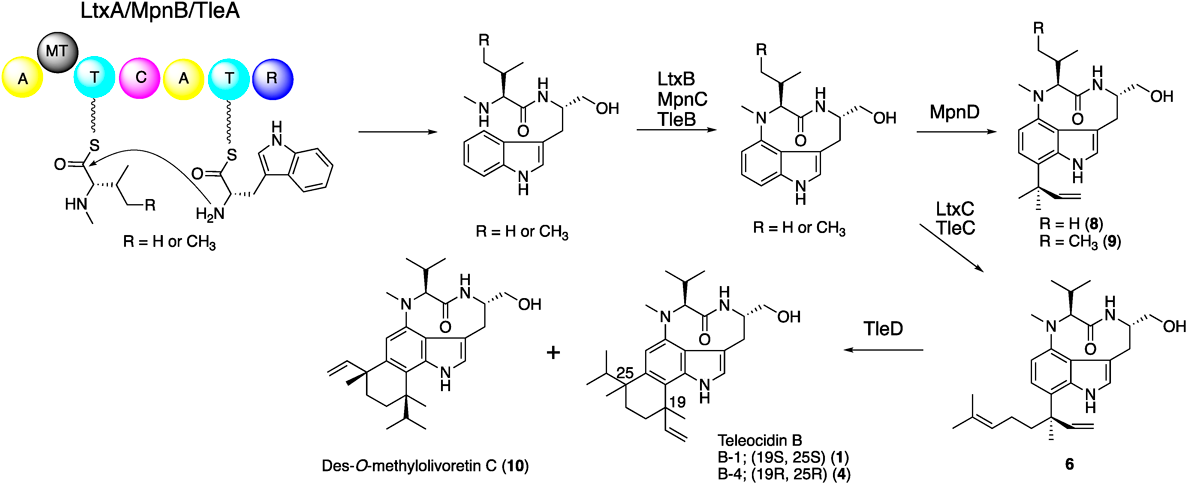
In 2012, Ju and colleagues reported the biosynthetic gene cluster (mpn cluster), responsible for the production of pendolmycin (8) and methylpendolmycin (9), from Marinactinospora thermotolerans SCSIO 00652.34) The mpn gene deletion experiments indicated that the NRPS MpnB, the CYP MpnC, and the prenyltransferase MpnD are sufficient to produce 8 and 9. The amino acid identities between LtxABC and MpnBCD are 40, 44, and 35%, respectively (Fig. 2). MpnB accepts both L-valine (Val) and L-isoleucine (Ile) to generate L-Val and L-Ile containing indolactam, respectively. While MpnD showed moderate sequence similarity to LtxC, this enzyme catalyzes the reverse prenylation of dimethyl allyl diphosphate (DMAPP) to the C7 position of the indole ring of indolactams to produce 8 and 9.
In 2014, Awakawa et al. identified and characterized the teleocidin Bs biosynthetic gene cluster (tle cluster) from Streptomyces blastmyceticus.35) The gene cluster contains the LtxABC homologs TleABC (48, 47, and 38%, amino acid identities, respectively) (Fig. 2). In addition to TleABC, the C-methyltransferase (C-MT) gene tleD was also identified outside of the tle cluster. The heterologous expression of tleABCD genes in S. lividans generated 1, 4, and des-O-methyl-olivoretin C (10), indicating that TleD performs the C-methylation and terpene cyclization reactions of 6.
In 2018, we functionally and structurally characterized TleB and its homologue HinD, identified from Streptoalloteichus hindustanus.36,37) HinD shares 58% amino acid sequence identity with TleB. HinD accepts both 7 and N-methyl-L-phenylalanyl-L-tryptophanol (NMFT, 11), while only 7 was accepted by TleB, and forms the C–N bond between the N13 atom and the C4 of indole to yield indolactam V (5) and indolactam F (12) (Fig. 3A). To elucidate the mechanism of the C–N bond formation reaction by P450 enzymes, the substrate analogues 13–17, in which the N13 atoms were substituted with various functional groups, including -OH, -OMe, and -NH2, were synthesized and used as substrates of TleB and HinD (Fig. 3B). The enzyme reactions of TleB and HinD revealed the generation of the 6/5/6 tricyclic compounds 18–22 from 13–17, respectively.
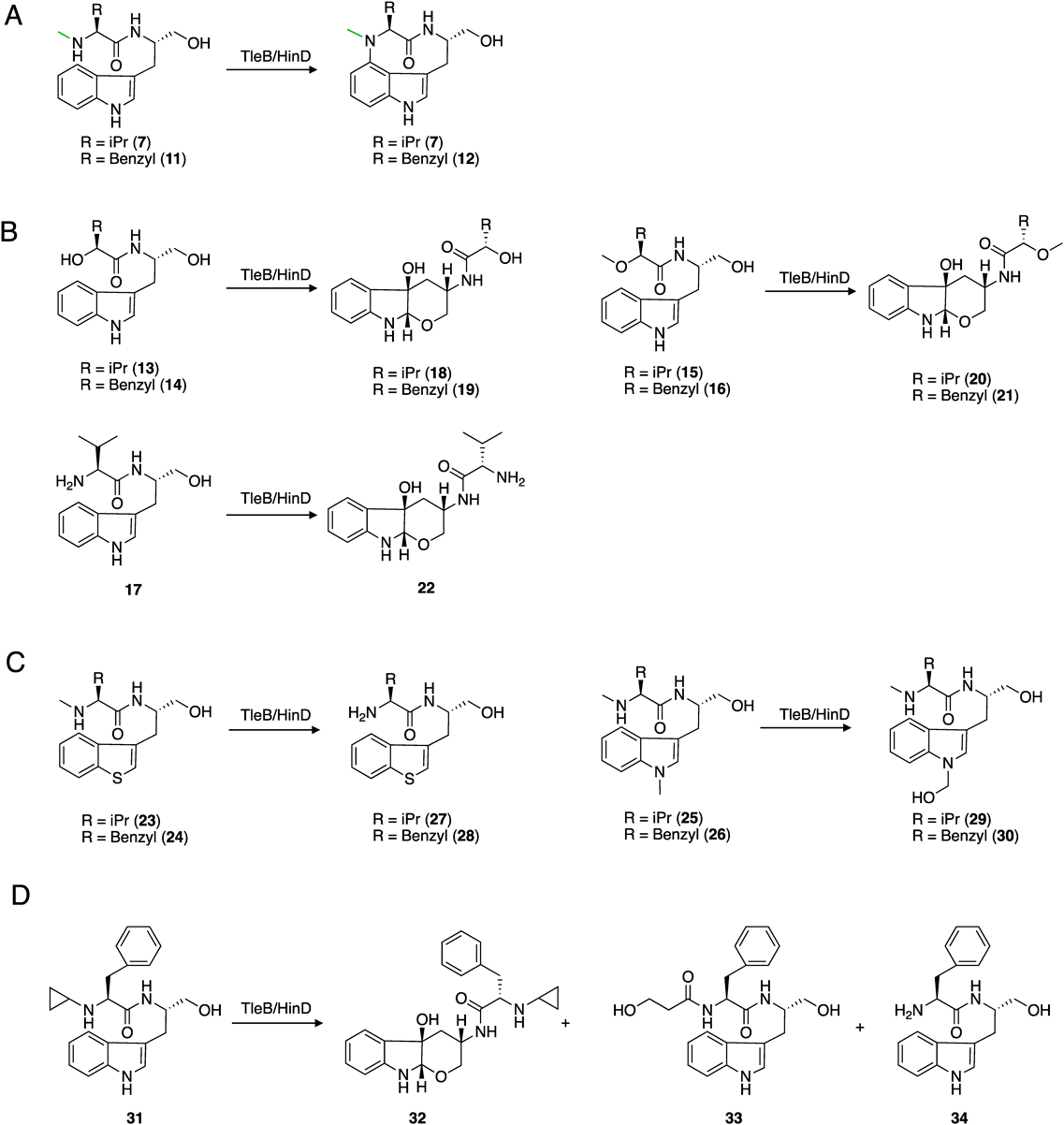
(A) The original reactions of TleB and HinD. (B) The reactions of TleB and HinD with 13th substituted substrate analogues. (C) The reactions of TleB and HinD with N1 substituted substrate analogues. (D) The reactions of TleB and HinD with an N13-cyclopropyl substituted analogue. (A) The original reactions of TleB and HinD. (B) The reactions of TleB and HinD with 13th substituted substrate analogues. (C) The reactions of TleB and HinD with N1 substituted substrate analogues. (D) The reactions of TleB and HinD with N13-cyclopropyl substituted analogue.
These compounds are generated through C–O bond formation between C2 and the hydroxyl group at C14, and hydroxylation at C3, suggesting that the 6/5/6 tricyclic scaffold is formed via a C2-C3-epoxide or 3β-hydroxy-indolenine intermediate, produced by hydrogen atom abstraction from the N1 position (Fig. 4). The intermediate is attacked by C14-OH to form the 6-membered rings of 18–22. Moreover, compounds 23–26, in which N1 position was substituted with sulfur atom or masked with methyl group, were synthesized and used as substrates. As a result, the enzymes produced 27–30 from 23–26, respectively, which are formed by the oxidation of the N13- or N1-methyl group (Fig. 3C). These results suggested that the N13 and indole N1 positions are the reactive points to generate radical species for the formation of the C–N bond. We also confirmed the generation of radical species on the N13 position by using the N13-cyclopropyl substituted analogue 31. The enzyme reaction of HinD with 31 produced the cyclopropane ring-opened compounds 32–34, indicating the generation of a radical on the N13 atom, and the following radical rearrangements open the cyclopropane ring (Fig. 3D). These enzyme reactions with substrate analogues suggested that 1) a hydrogen atom at N1 is first abstracted by the enzyme, 2) the radical is also generated on N13 position, and 3) the diradical coupling mechanism is likely for the enzymatic C–N bond formation reaction.

To elucidate the structural details of the C–N bond formation reaction, the structures of TleB with 7 and HinD with 11 and NO, which mimics the proximal oxygen atom during the reaction, were solved at 1.9 and 2.6 Å, respectively.37) The overall structures of TleB and HinD possess the trigonal prism-fold, consistent with other P450 enzymes (Figs. 5A, B). The binding sites of 7 and 11 in the active sites of TleB and HinD are similar and fixed by hydrogen bonding interactions, whereas the conformation of the dipeptide chain is different due to the substitution of the isopropyl moiety of 7 with the benzyl structure in 11 (Figs. 5C, D). The N1 atom of the substrate is close to the heme center in both cases. The distances between an iron-coordinated water molecule and N1 of 7 in TleB and between NO and N1 of 11 are 2.7 and 3.1 Å, respectively. These binding modes are consistent with the in vitro analysis, in which the hydrogen atom at N1 is initially abstracted by the heme center to generate the radical intermediate. In contrast, the distances between N13 and the iron-coordinated water molecule or NO are 6.7 Å (in TleB) and 8.2 Å (in HinD), which are too far to generate the second radical directly, and the distances between N1 and N13, 6.0 Å (in TleB) and 6.2 Å (in HinD), are also too far to react. Therefore, a conformational change of the radical intermediate is thought to occur to form the second radical on N13 and the C–N bond between N1 and N13. Intriguingly, the complex structure of HinD with 23 supports this hypothesis (Fig. 5E). In this structure, the C9/N10 and C11/C12 bonds in 23 are rotated 55° clockwise and 153° counterclockwise, respectively, as compared to those of 7, and the distances between an iron-coordinated water and N13 and between C4 and N13 are 3.1 and 3.8 Å, respectively. These distances are close enough for the hydrogen abstraction from N13 and the C–N bond formation. In the structures of TleB and HinD, the F287 residue is close to the indole ring of substrates, which may be important to stabilize the first radical on the intermediate. Indeed, the F287A variant exhibited dramatically reduced activity. Based on these results, it is likely that the substrate undergoes a conformational change after the initial hydrogen abstraction from N1 to generate the second radical, and subsequently the C–N bond is formed between C4 and N13 through a radical coupling reaction (Fig. 6).
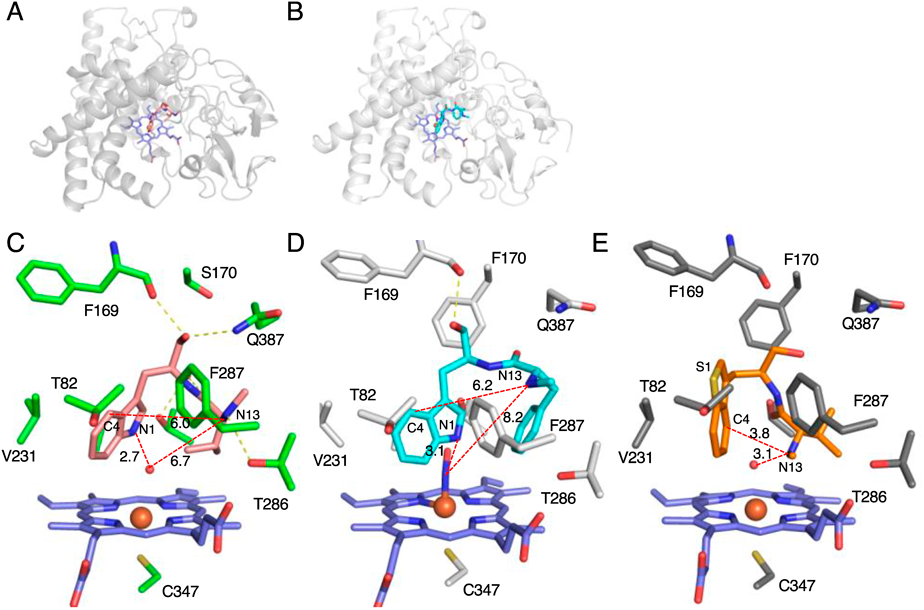
(A, B) The overall structures of (A) TleB and (B) HinD. (C) The active site of the complex structure of TleB with 7. (D) The active site of the complex structure of HinD with 11. (E) The active site of the complex structure of HinD with 23. The substrates and heme molecules are depicted by stick models. Dashed yellow lines represent hydrogen bonds. Dashed red lines represent the distances between key atoms. Water molecules are shown as red nb_spheres.

The in vitro and structural analyses of TleB and HinD suggested that the dissociation energy of the X–H bond and the stability of the radical on the heteroatom are important for the formation of the nine-membered ring. Therefore, we assumed that a nine-membered ring can be formed when the N13-methyl group of 7 is substituted with a thiol group, because the dissociation energy of S–H is lower than that of NMe–H and the thiyl radical is more stable than the N-methyl radical. To test this hypothesis, we synthesized the substrate analogue 35, which has a thiol group at the 13th position, and used it as a substrate of TleB. As a result, the enzyme generated two enzyme reaction products, 36 and 3738) (Fig. 7). The structural analysis of the enzyme reaction products revealed that 36 is a novel sulfur atom-containing indolactam derivative, thioindolactam V (36). Interestingly, conformational analyses of 36 by NMR and X-ray studies, as well as density functional theory (DFT) calculations, indicated that this compound only exists in the so-called “SOFA” conformation, which is derived from the trans-lactam amide (Fig. 8). In contrast, 5 is known to exist as a mixture of two stable conformations, derived from the cis-lactam amide “TWIST” and the trans-lactam amide “SOFA” in the solution state (TWIST: SOFA = 2 : 1).39)
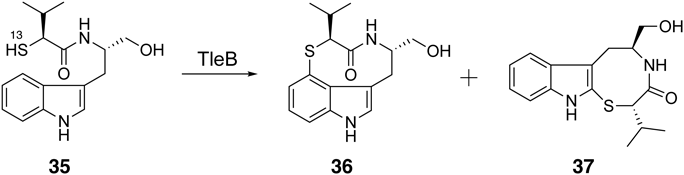
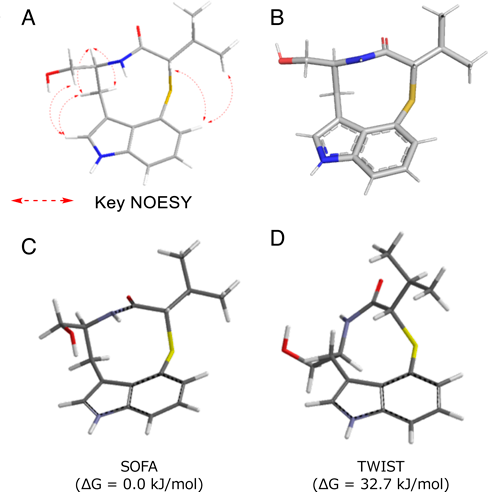
(A) NMR analysis. (B) Single X-ray crystal analysis, and (C) DFT calculation analysis.
Using the crystalline sponge method,40) the structure of product 37 was determined to be a novel 6/5/8-tricyclic compound, which is generated via C–S bond formation between C2 and S13. Notably, the hydroxy group on C3 was eliminated in compound 37, in contrast to the 6/5/6 tricyclic compounds 18–22. This would be due to the different conformation of the cyclized intermediate (Fig. 9). The mechanism for the production of 37 was proposed as follows. The hydrogen atom abstraction from N1 and the following hydroxyl rebound reaction generate a 3β-hydroxy-indolenine (or 2,3-epoxide) intermediate as in the case of 18–22 formation reactions (Fig. 9A). Subsequently, the intramolecular attack of the thiol group at the 13th position from the re-face generates the 2R, 3R cyclized intermediate, which is followed by water elimination and re-aromatization of the indole ring to yield 37. On the other hand, the water elimination is not occurred in 18–22 formation reactions because the 14-OH in 13–17 can attack only from the si-face of the indole C2 and produce the 2S, 3R cyclized intermediate (Fig. 9B).

(A) The formation and dehydration of 37. (B) The cyclization mechanisms of the 6/5/6 product.
The in vitro analyses of TleC and MpnD with different lengths (C5–C25) of prenyl substrates revealed that these enzymes show broad substrate specificity against the length of the prenyl donor, while TleC and MpnD prefer C10 and C5, respectively41) (Fig. 10). To elucidate the molecular basis for the reverse prenylation on the C7 position of the indole ring, the apo and complex structures of TleC and MpnD with the DMAPP analogue dimethylallyl S-thio-pyrophosphate (DMSPP) and 7 were solved in 2016.41) The overall structures of these enzymes share the αββα barrel fold, as observed in other aromatic prenyltransferases (Figs. 11A, B). The comparison of the active site architectures between the complex structures of TleC and MpnD indicated that three residues, Trp97, Phe170, and Ala173, in TleC are substituted with Tyr80, Trp157, and Met159, respectively, in MpnD (Figs. 11C, D). In the complex structure of TleC, a large pocket for binding the prenyl side chain of GPP (C10) is observed, whereas this pocket is eliminated by the small-to-large substitutions of Phe170/Ala173 with Trp157/Met159 in the active site of MpnD. These observations suggested that this prenyl binding pocket controls the preference of the prenyl chain length of substrates and the regio-selectivity of prenylation reactions.

(A, B) The overall structures of (A) TleC and (B) MpnD. (C) The active site of the complex structure of TleC with 5 and DMSPP. (D) The active site of the complex structure of MpnD with 5 and DMSPP.
To control the substrate specificities of the TleC and MpnD prenyltransferases, we conducted structure-based engineering.41) The substitution of Ala173 in TleC with Met altered the preference for the prenyl substrate length. The TleC A173M variant efficiently used C5 DMAPP instead of C10 GPP to yield 9 (Fig. 12A). In contrast, the MpnD M159A variant preferred C10 GPP as a prenyl donor instead of C5 GPP to produce 6 (Fig. 12B). Furthermore, the TleC W97Y/A173M and W97Y/F170W/A173M variants obtained a new function to generate teleocidin A-2 (38),10) the C19 epimer of 6 as the major product, together with 6 and 5-geranylindolactam V (39) (Fig. 12C). These results support the hypothesis that these three residues, which form the prenyl binding pocket, are important for the selectivity of the prenyl chain length and the regio- and stereo-selectivities of the prenylation reactions.

The in vitro analysis of the methyltransferase TleD with 6 and S-adenosylmethionine (SAM) generated the same products 1, 4, and 10 with same ratio (1 : 4 : 10 = 1 : 5 : 2) as the heterologous expression of tleABCD, suggesting that the TleD-catalyzed methylation reaction triggers the terpene cyclization reaction.35) Furthermore, the enzyme reaction of TleD with 25-D-labeled 6 reveled the migration of the deuterium atom from C25 to C26, suggesting the formation of the spiro-ring containing intermediate. Based on the in vitro enzyme reaction and the deuterium-label experiment, the mechanism of the methylation-mediated cyclization reaction by TleD was proposed as follows (Fig. 13). First, C25 of 6 is methylated and a cation is generated on C26. Then, the 1,2-hydride shift from C25 to C26 transfers the cation to C25. Subsequent nucleophilic attacks by C7 of the indole ring to the C25 cation from the Si-face or Re-face form the spiro-ring-containing intermediates A and B, respectively. The carbon rearrangement of the spiro-ring moiety of intermediate A leads to the generation of 1 via path a. In contrast, the C–C bond rearrangements of intermediate B produce 4 through path b, while 10 is yielded via path c. Here, the steric hindrance from the bulky isopropyl and vinyl groups would determine the ratio of products (1 : 4 : 10 = 1 : 5 : 2). Since no intermediates or shunt products were observed in the reaction, the rearrangement reactions would proceed spontaneously after the formation of the C26 cation intermediate.

The complex structure of TleD with substrate 6 and S-adenosyl homocysteine (SAH) was solved at 2.8 Å resolution.42) TleD forms a homohexamer, and the overall structure of TleD has a typical class I SAM-dependent methyltransferase fold with an additional N-terminal α-helix (Figs. 14A, B). The SAH is bound at the dimer surface in the Rossmann fold, through many hydrogen bonds and van der Waals interactions (Fig. 14C). The substrate 6 is bound in a hydrophobic cavity and the orientation of 6 would be determined by the hydrogen bond interactions with active site residues, such as Glu153 and Glu181. The distance between Sδ of SAH and C25 of 6 is 4.5 Å, which is reasonable for the methylation reaction. The molecular dynamics simulation of TleD and substrates with different geranyl group conformations suggested that the preferred dihedral angle of C23–C24–C25–C26 is close to 60–90°. In this conformation, the distance between methyl group of SAM and C25 of 6 is maintained at less than 4 Å, which supports the first methylation step of the proposed enzyme reaction of TleD. However, the structural bases for the following 1,2-hydride shift and the C–C bond rearrangement of the spiro-intermediate to produce 1, 4, and 10 remain unclear. To understand the mechanism in more detail, further calculations and molecular dynamics studies are required.

(A, B) Overall structures of (A) the TleD homohexamer and (B) the TleD dimer. (C) The active site of the complex structure of TleD with 6 and SAH. Dashed yellow lines represent hydrogen bonds. Dashed red lines represent distances between key atoms.
This review has summarized the investigations on the functions, structures, mechanisms, and enzyme engineering of teleocidin biosynthetic enzymes. Based on the in vitro enzymatic analysis and the X-ray crystal structure, the detailed reaction mechanisms of the C–N bond formation by the P450 enzyme TleB, the reverse prenylation by the prenyltransferase TleC, and the methylation-mediated cyclization reaction by the methyltransferase TleD have been proposed. However, there are still some unresolved issues in these reactions, such as the conformational change of the radical intermediate in the TleB reaction and the C–C bond rearrangement of the spiro-ring intermediate in the TleD reaction. Future computational studies, including DFT calculations, docking simulations of intermediates, and molecular dynamics, of these enzymes will provide important insights into the detailed reaction mechanisms.
Enzyme engineering studies of teleocidin biosynthetic enzymes have produced various teleocidin and indolactam derivatives. These results also shed light on the value of rational manipulation of enzymes to generate structurally diverse unnatural products. The future applications of such biosynthetic enzymes as biocatalysts will provide medicinally important compounds with unnatural and novel scaffolds for drug discovery.
The authors would like to express sincere appreciation to an excellent group of coworkers whose contributions are cited in the text. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (JSPS KAKENHI Grant Number 19K15703), the Takeda Science Foundation, and the PRESTO program from Japan Science and Technology Agency (JPMJPR20DA).
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2022 Pharmaceutical Society of Japan Award for Young Scientists.