Abstract
The capsid of human immunodeficiency virus type 1 (HIV-1) forms a conical structure by assembling oligomers of capsid (CA) proteins and is a virion shell that encapsulates viral RNA. The inhibition of the CA function could be an appropriate target for suppression of HIV-1 replication because the CA proteins are highly conserved among many strains of HIV-1, and the drug targeting CA, lenacapavir, has been clinically developed by Gilead Sciences, Inc. Interface hydrophobic interactions between two CA molecules via the Trp184 and Met185 residues in the CA sequence are indispensable for conformational stabilization of the CA multimer. Our continuous studies found two types of small molecules with different scaffolds, MKN-1 and MKN-3, designed by in silico screening as a dipeptide mimic of Trp184 and Met185 have significant anti-HIV-1 activity. In the present study, MKN-1 derivatives have been designed and synthesized. Their structure–activity relationship studies found some compounds having potent anti-HIV activity. The present results should be useful in the design of novel CA-targeting molecules with anti-HIV activity.
Introduction
Because the pandemic of the novel coronavirus disease 2019 (COVID-19), which is caused by a positive-strand RNA virus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has threatened our daily lives more than three years, conquest of viral infectious diseases is a major task for human beings.1) Human immunodeficiency virus type 1 (HIV-1) is classified as a member of a retrovirus family, and causes AIDS. At present, inhibitors of reverse transcriptase,2) protease3) and integrase,4,5) in combination antiretroviral therapy (cART) are clinically used for the chemotherapy of HIV infectious diseases.6–8) cART has some drawbacks however, including the emergence of mutant strains with multi-drug resistance, the appearance of serious side effects and the cost of the resulting drugs. Therefore, we have continued enormous efforts to resolve these issues and increase the alternatives and repertoires of anti-HIV-1 drugs, and sought anti-HIV agents with various mechanisms of action such as co-receptor CXCR4 antagonists,9–15) CD4 mimics,16–21) fusion inhibitors,22–25) integrase inhibitors26–28) and inhibitors of viral uncoating and viral assembly.29–33) In the present study we focused on inhibitors of viral uncoating and viral assembly29–33) among these attractive targets.
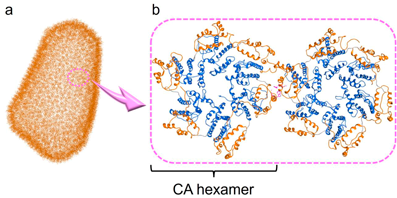
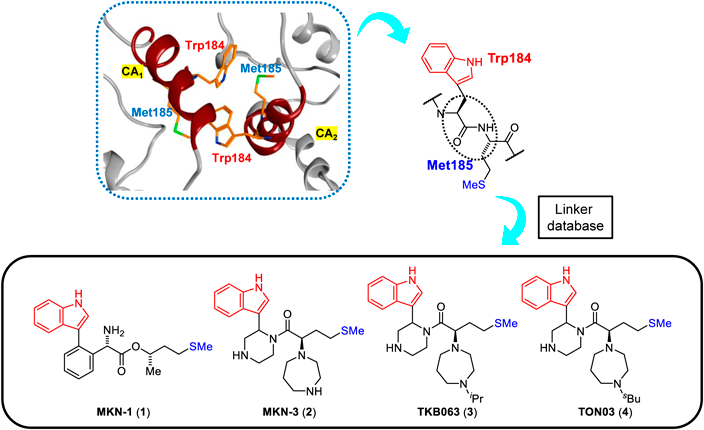
The Gag precursor protein of HIV, Pr55Gag, produces the capsid (CA) protein, which consists of N- and C-terminal domains (NTD/CTD) and is highly conserved among various strains.34,35) Assembly through oligomerization of hexamers and pentamers of the CA molecules36) forms a CA core with a conical structure,37,38) which encapsulates the RNA genome, the reverse transcriptase and the integrase. In addition, Pr55Gag expresses the matrix (MA) protein, which is correlated to the formation of the virion shell39,40) The MA and CA proteins might be rationale targets for suppression of viral replication. Accordingly, several MA and CA fragment derivatives have been found to have significant anti-HIV activity in our29–33) and other laboratories.41–44) Since the uncoating/assembly driven by the degradation/oligomerization of the MA and CA proteins is performed inside host cells, these peptide fragments must penetrate cell membranes to attack the MA and CA proteins. Therefore, an octa-arginyl moiety45) was added to the peptide-derived inhibitors to enhance their cell membrane permeability.29–33) Since small hydrophobic molecules might penetrate cell membranes by themselves without an octa-arginyl moiety, small compounds with inhibitory activity against viral uncoating and assembly are desirable. Several small compounds targeting the CA proteins have been discovered to date,46–63) and among them the drug, lenacapavir, has been clinically developed by Gilead Sciences, Inc.59) Since this target has been proven to be valid, drug repertoires are required. The dimer of HIV-1 CA proteins has a hydrophobic interaction through Trp184 in Helix 9 of CTD of one CA molecule and Met185 of the other molecule in the structural analysis.36–38,64) This interaction is indispensable for conformational stabilization of the CA hexamers and pentamers for assembly to form the CA core (Fig. 1). Viral mutant with Trp184Ala and Met185Ala mutations has very weak dimeric interactions between two CA molecules and shows an abnormal morphology of the viral particles with no infectivity.65) Trp184 and Met185 are highly conserved among natural HIV/simian immunodeficiency virus (SIV) strains.20) In our previous studies on search for anti-HIV peptides from CA fragment derivatives, the peptide that includes Trp184 and Met185 has significant anti-HIV activity.66)
In our design of small compounds, the site of this hydrophobic interaction through Trp184 of one molecule and Met185 of a second molecule might be a valid target for CA dysfunction. Using in silico screening we designed small compounds that might bind to the hydrophobic interaction site.67) A series of dipeptide mimics of Trp184 and Met185 were constructed using the Molecular Operating Environment (MOE) (Chemical Computing Group Inc., Montreal, Quebec, Canada). Briefly, in the CA dimer, the structure of the first monomer molecule (CA1) of the CA protein dimer (PDB ID: 3J34) remained as a receptor, and the backbone structure of Trp184 and Met185 of a second monomer molecule (CA2) was removed. The side-chain structures of these two residues of the second monomer molecule (CA2) remained in place, and the scaffold structures crosslinking two side-chain functional groups were screened based on the linker database provided by the Scaffold Replacement application of MOE to bind to the above receptor side of the first monomer molecule (CA1) (Fig. 2). Screened scaffold structures of dipeptide mimics were selected based on the following scores which include binding affinity for receptors, ligand efficacy (London dG), topological polar surface area, molecular weight, log of the octanol/water partition coefficient (SlogP), and an estimate of the feasibility of synthetic access.68) London dG values show binding affinities for the target protein; smaller values mean higher binding affinities, and compounds with values of <−6 have higher binding affinity for the receptor side of the first CA monomer molecule (CA1) when compared to the interaction between two CA molecules because the London dG value of the CA dimer is approximately −6. This screening strategy provided some candidates with high binding affinity, including MKN-1 (1) and MKN-3 (2) with London dG values of −9.134 and −9.555, respectively.67,69) MKN-1 (1) and MKN-3 (2) derivatives, TKB063 (3) and TON03 (4), showed significant anti-HIV activity at the level of micromolar,67,69) although MKN-3 (2) itself failed to show anti-HIV activity below 50 µM because of relatively high hydrophilicity of the compound.69) In the present study, several derivatives of MKN-1 (1) were synthesized, and their anti-HIV activity and cytotoxicity were evaluated.
Results and Discussion
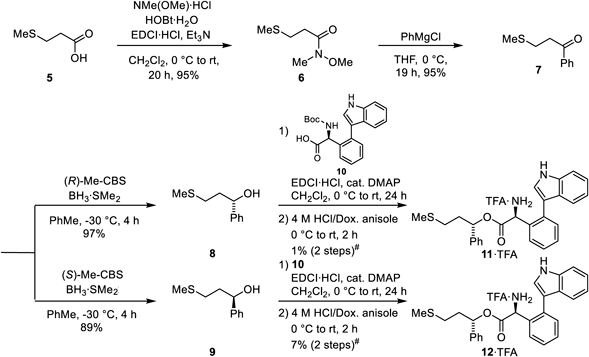
Chemistry. Compounds 11 and 12Treatment of the Weinreb amide (6) of carboxylic acid (5) with phenylmagnesium chloride gave the ketone (7) followed by the Corey–Bakshi–Shibata (CBS) Reduction with borane and chiral oxazaborolidines as catalyst70) to yield separately alcohols 8 and 971) (Chart 1). The subsequent esterification of 8 and 9 with Nα-tert-butoxycarbonyl (Boc)-protected indole-containing phenylglycine (10)67) followed by deprotection of the Nα-Boc group yielded esters (11) and (12), respectively. Esters (11) and (12) showed relatively low yields because of low recovery after purification of the target compounds by HPLC.
Compounds 18–21Treatment of aldehyde 13 with several Grignard reagents, ethylmagnesium chloride, tert-butylmagnesium chloride, iso-propylmagnesium chloride and n-pentylmagnesium chloride, gave the corresponding alcohols (14–17), respectively (Chart 2). The subsequent esterification of 14–17 with Nα-Boc-protected indole-containing phenylglycine (10)67) followed by deprotection of the Nα-Boc group yielded esters (18–21), respectively, as diastereomixtures. Esters (18–21) showed relatively low yields because of low recovery after HPLC purification of the target compounds.

Chart 2. Synthesis of Compounds 18–21
Treatment of diol 22 with sodium hydride and methyl iodide gave ether 23 (Chart 3). The subsequent esterification of 23 with Nα-Boc-protected indole-containing phenylglycine (10)67) followed by deprotection of the Nα-Boc group yielded the ester (24).

Chart 3. Synthesis of Compound 24
The esterification of 25 with Nα-Boc-protected indole-containing phenylglycine (10)67) followed by deprotection of the Nα-Boc group yielded MKU-010 (26) (Chart 4).

Chart 4. Synthesis of MKU-010 (26)
The amidation of MKN-1 (1) with acids 27–29 gave the amides (30–32) (Chart 5). Compounds 31 and 32 showed relatively low yields because of low recovery of the target compounds on HPLC purification.
Evaluation of Anti-HIV Activity and CytotoxicityThe anti-HIV activity of the synthesized compounds was assessed based on their protection against HIV-1 (NL4-3 strain)-induced cytopathogenicity in MT-4 cells using an 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay.29–33) The cytotoxicity of these compounds was determined based on reduction of the viability of MT-4 cells using an MTT assay. These results are shown in Table 1. In our previous study, although MKN-3 (2), with a London dG value of −9.555, has high binding affinity for a CA molecule, it failed to show significant anti-HIV activity, possibly due to the poor cell membrane permeability of MKN-3 (2), which therefore could not penetrate the cells.69) In general, the hydrophobicity of compounds is correlated to their cell membrane permeability.73) The Log P value of MKN-3 (2) is 0.27, which is markedly lower than that of MKN-1 (1) (LogP: 2.97), which show significant anti-HIV activity.67) As a result, hydrophobic derivatives of MKN-3 (2), TKB063 (3) and TON03 (4), showed significant anti-HIV activity.69) Therefore, Log P values of compounds are considered and shown in Table 1.
Table 1. Anti-HIV Activity, Cytotoxicity and Log
p Values of MKN-1 (
1) Derivatives
aEC50 values are the concentrations for 50% protection from HIV-1 (NL4-3 strain)-induced cytopathogenicity in MT-4 cells. bCC50 values are the concentrations for 50% reduction of the viability of MT-4 cells. The data are a mean value of at least two independent experiments. cCalculated using ChemDraw Professional 15.1. ddiastereomixture.
Compounds 11, 12, and 18–21 are derivatives of MKN-1 (1) with replacement of the methyl group by several substituents. All these compounds showed significant decreases of anti-HIV activity from MKN-1 (1). Compound 21 failed to show anti-HIV activity below 25 µM. Since Log P values of these compounds are 3.4–4.7, the hydrophobicity of compounds is appropriate. Since compounds 11 and 12 have almost the same potency, the bulky phenyl group fails to fit in the pocket. Therefore, relatively small substituents such as a methyl group are suitable to fit in the pocket and express high anti-HIV activity. All these compounds showed almost the same levels of cytotoxicity (CC50: 20–35 µM), suggesting modest cytotoxicity.
Compounds 24 and MKU-010 (26) are derivatives of MKN-1 (1) with replacement of the sulfur atom of the methylsulfide moiety by oxygen and methylene, respectively. MKU-010 (26) increased slightly the anti-HIV activity of MKN-1 (1), while compound 24 decreased remarkably the anti-HIV activity of MKN-1 (1). In general, sulfur and oxygen atoms have common characteristic features and can often be changed each other without any effect on its activity. This is not true of the current case. On the contrary, a methylene unit is more suitable surrogate of a sulfur atom than an oxygen atom. It suggests that the methylsulfide moiety interacts with the hydrophobic interface of the target CA molecule involving Trp184 and Leu189 and that a methyl ether group is not suitable for hydrophobic interactions due to its hydrophilicity while an alkyl group is suitable for hydrophobic interactions. The relatively high hydrophilicity of the whole compound of 24 (Log P value: 2.25) might be the reason for low anti-HIV activity. MKU-010 (26) has proper hydrophobicity (Log P value: 3.96). MKU-010 (26) increased cytotoxicity of MKN-1 (1), while compound 24 decreased cytotoxicity of MKN-1 (1).
Compounds 30–32 are derivatives of MKN-1 (1) with an additional sulfide or indole moiety at free amino group. All these compounds failed to show significant anti-HIV activity below 20 µM or the CC50 value, although these compounds have appropriate hydrophobicity (Log P value: 3.2–4.4). Compounds 30–32 showed significant cytotoxicity. It indicates that modification by an additional sulfide or indole moiety at free amino group is not suitable.
The docking simulation of MKN-1 (1) and a CA protein (PDB ID: 3J34) operated by Molecular Operating Environment (MOE), 2022.02.73) disclosed that the amino group of MKN-1 (1) and the carboxylate group of Glu175 in the CA protein are closely located and form an ionic interaction each other (Fig. 3). Since the amino group of compounds 30–32 is protected with an additional sulfide or indole moiety, these compounds might fail to form an ionic interaction with the carboxylate group of Glu175. Therefore, these compounds fail to show significant anti-HIV activity.
Conclusion
MKN-1 (1) derivatives were developed based on in silico screening as a dipeptide mimic of Trp184 and Met185 at the hydrophobic interaction site between two CA molecules that are important for stabilization of the oligomeric structure of CA proteins. In this study, a novel compound with higher anti-HIV activity than that of the original compound MKN-1 (1) was found: MKU-010 (26) with replacement of the sulfur atom of the methylsulfide moiety by methylene increased slightly the anti-HIV activity of MKN-1 (1). It suggests that the methylsulfide moiety interacts with the hydrophobic interface of the target CA molecule because an alkyl group is suitable for hydrophobic interactions. The proper hydrophobicity of MKU-010 (26) (Log P value: 3.96) might be the reason for high anti-HIV activity. The present study brought useful information that small substituents such as a methyl group as a branched group of the alkyl chain next to the ester moiety of MKN-1 (1) are suitable to fit in the pocket of the target CA molecule and express high anti-HIV activity. In addition, the amino group of MKN-1 (1) should be free for expression of high anti-HIV activity because this amino group forms an ionic interaction with the carboxylate group of Glu175. Development of a new class of small molecules of anti-HIV agents would be desired in conjugation with MKN-3 (2) derivatives, which have a scaffold structure different from that of MKN-1 (1) derivatives. Recently, we developed anti-hepatitis B virus (HBV) agents targeting an HBV capsid protein (HBc).74) Therefore, capsid proteins are great therapeutical targets against hepatitis B as well as HIV infectious diseases. The present data will be useful and important in the future design of new anti-HIV agents targeting HIV-1 CA proteins.
Experimental
Compound SynthesisThe synthetic methods for target compounds 11, 12, 18–21, 24, MKU-010 (26) and 30–32 are described in Charts 1–3. The purity of all the final compounds measured by analytical reverse phase (RP)-HPLC or NMR is >95%. Experimental procedures including characterization data are provided in the Supplementary Materials.
Evaluation of Anti-HIV-1 Activity and CytotoxicityExperimental procedures are provided in the Supplementary Materials.67,69,75)
Acknowledgments
This work was supported in part by the Grant for Research Program on HIV/AIDS, Japan Agency for Medical Research and Development (AMED) under Grant Number 18fk0410004 (T. M., H. S. and H. T.) and JP23fk041004 (T. K. and M. Y.); JSPS KAKENHI Grant Number 20H03362 (H. T.), 22K08615 (T. M. and H. T.), 22K15243 (K. T.), and 23K14318 (T. K.); AMED under Grant Number JP23ama121043 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) (H. T.); JPMJFS2109 (MEXT, the establishment of university fellowships towards the creation of science technology innovation) (T. I. and Y. M.). This research is based on the Cooperative Research Project of Research Center for Biomedical Engineering.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials. Supplementary Materials in PDF format contain experimental procedures and compound characterization data (1H-NMR, 13C-NMR, and HRMS).
References
- 1) Mitsuya H., Kokudo N., Glob. Health Med., 2, 53–55 (2020).
- 2) Mitsuya H., Weinhold K. J., Furman P. A., St Clair M. H., Lehrman S. N., Gallo R. C., Bolognesi D., Barry D. W., Broder S., Proc. Natl. Acad. Sci. U.S.A., 82, 7096–7100 (1985).
- 3) Ghosh A. K., Dawson Z. L., Mitsuya H., Bioorg. Med. Chem., 15, 7576–7580 (2007).
- 4) Cahn P., Sued O., Lancet, 369, 1235–1236 (2007).
- 5) Grinsztejn B., Nguyen B.-Y., Katlama C., Gatell J. M., Lazzarin A., Vittecoq D., Gonzalez C. J., Chen J., Harvey C. M., Isaacs R. D., Lancet, 369, 1261–1269 (2007).
- 6) Ohashi N., Tamamura H., “Amino Acids, Peptides and Proteins,” Vol. 41, Chap. 1, ed. by Ryadnov M., Hudecz F., The Royal Society of Chemistry, Cambridge, 2017, pp. 1–29.
- 7) Tamamura H., Kobayakawa T., Ohashi N., “Mid-size drugs based on peptides and peptidomimetics: A New Drug Category,” Springer Briefs in Pharmaceutical Science & Drug Development, Singapore, 2018.
- 8) Maeda K., Das D., Kobayakawa T., Tamamura H., Takeuchi H., Curr. Top. Med. Chem., 19, 1621–1649 (2019).
- 9) Murakami T., Nakajima T., Koyanagi N., Tachibana K., Fujii N., Tamamura H., Yoshida N., Waki M., Matsumoto A., Yoshie O., Kishimoto T., Yamamoto N., Nagasawa T., J. Exp. Med., 186, 1389–1393 (1997).
- 10) Tamamura H., Xu Y. O., Hattori T., Zhang X. Y., Arakaki R., Kanbara K., Omagari A., Otaka A., Ibuka T., Yamamoto N., Nakashima H., Fujii N., Biochem. Biophys. Res. Commun., 253, 877–882 (1998).
- 11) Fujii N., Oishi S., Hiramatsu K., Araki T., Ueda S., Tamamura H., Otaka A., Kusano S., Terakubo S., Nakashima H., Broach J. A., Trent J. O., Wang Z. X., Peiper S. C., Angew. Chem. Int. Ed., 42, 3251–3253 (2003).
- 12) Tamamura H., Hiramatsu K., Mizumoto M., Ueda S., Kusano S., Terakubo S., Akamatsu M., Yamamoto N., Trent J. O., Wang Z. X., Peiper S. C., Nakashima H., Otaka A., Fujii N., Org. Biomol. Chem., 1, 3663–3669 (2003).
- 13) Tanaka T., Nomura W., Narumi T., Masuda A., Tamamura H., J. Am. Chem. Soc., 132, 15899–15901 (2010).
- 14) Tanaka T., Narumi T., Ozaki T., Sohma A., Ohashi N., Hashimoto C., Itotani K., Nomura W., Murakami T., Yamamoto N., Tamamura H., ChemMedChem, 6, 834–839 (2011).
- 15) Sakyiamah M. M., Kobayakawa T., Fujino M., Konno M., Narumi T., Tanaka T., Nomura W., Yamamoto N., Murakami T., Tamamura H., Bioorg. Med. Chem., 27, 1130–1138 (2019).
- 16) Yamada Y., Ochiai C., Yoshimura K., Tanaka T., Ohashi N., Narumi T., Nomura W., Harada S., Matsushita S., Tamamura H., Bioorg. Med. Chem. Lett., 20, 354–358 (2010).
- 17) Yoshimura K., Harada S., Shibata J., Hatada M., Yamada Y., Ochiai C., Tamamura H., Matsushita S., J. Virol., 84, 7558–7568 (2010).
- 18) Mizuguchi T., Harada S., Miura T., Ohashi N., Narumi T., Mori H., Irahara Y., Yamada Y., Nomura W., Matsushita S., Yoshimura K., Tamamura H., Bioorg. Med. Chem. Lett., 26, 397–400 (2016).
- 19) Ohashi N., Harada S., Mizuguchi T., Irahara Y., Yamada Y., Kotani M., Nomura W., Matsushita S., Yoshimura K., Tamamura H., ChemMedChem, 11, 940–946 (2016).
- 20) Kobayakawa T., Konno K., Ohashi N., Takahashi K., Masuda A., Yoshimura K., Harada S., Tamamura H., Bioorg. Med. Chem. Lett., 29, 719–723 (2019).
- 21) Kobayakawa T., Tsuji K., Konno K., Himeno A., Masuda A., Yang T., Takahashi K., Ishida Y., Ohashi N., Kuwata T., Matsumoto K., Yoshimura K., Sakawaki H., Miura T., Harada S., Matsushita S., Tamamura H., J. Med. Chem., 64, 1481–1496 (2021).
- 22) Otaka A., Nakamura M., Nameki D., Kodama E., Uchiyama S., Nakamura S., Nakano H., Tamamura H., Kobayashi Y., Matsuoka M., Fujii N., Angew. Chem. Int. Ed., 41, 2937–2940 (2002).
- 23) Nomura W., Hashimoto C., Ohya A., Miyauchi K., Urano E., Tanaka T., Narumi T., Nakahara T., Komano J. A., Yamamoto N., Tamamura H., ChemMedChem, 7, 205–208 (2012).
- 24) Nomura W., Hashimoto C., Suzuki T., Ohashi N., Fujino M., Murakami T., Yamamoto N., Tamamura H., Bioorg. Med. Chem., 21, 4452–4458 (2013).
- 25) Kobayakawa T., Ebihara K., Honda Y., Fujino M., Nomura W., Yamamoto N., Murakami T., Tamamura H., ChemBioChem, 20, 2101–2108 (2019).
- 26) Suzuki S., Urano E., Hashimoto C., et al., J. Med. Chem., 53, 5356–5360 (2010).
- 27) Suzuki S., Maddali K., Hashimoto C., Urano E., Ohashi N., Tanaka T., Ozaki T., Arai H., Tsutsumi H., Narumi T., Nomura W., Yamamoto N., Pommier Y., Komano J. A., Tamamura H., Bioorg. Med. Chem., 18, 6771–6775 (2010).
- 28) Nomura W., Aikawa H., Ohashi N., Urano E., Metifiot M., Fujino M., Maddali K., Ozaki T., Nozue A., Narumi T., Hashimoto C., Tanaka T., Pommier Y., Yamamoto N., Komano J. A., Murakami T., Tamamura H., ACS Chem. Biol., 8, 2235–2244 (2013).
- 29) Narumi T., Komoriya M., Hashimoto C., Wu H., Nomura W., Suzuki S., Tanaka T., Chiba J., Yamamoto N., Murakami T., Tamamura H., Bioorg. Med. Chem., 20, 1468–1474 (2012).
- 30) Mizuguchi T., Ohashi N., Nomura W., Komoriya M., Hashimoto C., Yamamoto N., Murakami T., Tamamura H., Bioorg. Med. Chem., 23, 4423–4427 (2015).
- 31) Mizuguchi T., Ohashi N., Matsumoto D., Hashimoto C., Nomura W., Yamamoto N., Murakami T., Tamamura H., Biopolymers: Peptide Science., 108, e22920 (2017).
- 32) Tsuji K., Owusu K. B.-A., Kobayakawa T., Wang R., Fujino M., Kaneko M., Yamamoto N., Murakami T., Tamamura H., Bioorg. Med. Chem., 28, 115488 (2020).
- 33) Tsuji K., Wang R., Kobayakawa T., Owusu K. B.-A., Fujino M., Kaneko M., Yamamoto N., Murakami T., Tamamura H., Bioorg. Med. Chem., 30, 115923 (2021).
- 34) Sarngadharan M. G., Bruch L., Popovic M., Gallo R. C., Proc. Natl. Acad. Sci. U.S.A., 82, 3481–3484 (1985).
- 35) Mervis R. J., Ahmad N., Lillehoj E. P., Raum M. G., Salazar F. H. R., Chan H. W., Venkatesan S., J. Virol., 62, 3993–4002 (1988).
- 36) Pornillos O., Ganser-Pornillos B. K., Kelly B. N., Hua Y., Whitby F. G., Stout C. D., Sundquist W. I., Hill C. P., Yeager M., Cell, 137, 1282–1292 (2009).
- 37) Ganser B. K., Li S., Klishko V. Y., Finch J. T., Sundquist W. I., Science, 283, 80–83 (1999).
- 38) Pornillos O., Ganser-Pornillos B. K., Yeager M., Nature (London), 469, 424–427 (2011).
- 39) Freed E. O., Virology, 251, 1–15 (1998).
- 40) Bukrinskaya A., Virus Res., 124, 1–11 (2007).
- 41) Zentner I., Sierra L.-J., Maciunas L., Vinnik A., Fedichev P., Mankowski M. K., Ptak R. G., Martín-García J., Cocklin S., Bioorg. Med. Chem. Lett., 23, 1132–1135 (2013).
- 42) Niedrig M., Gelderblom H. R., Pauli G., März J., Bickhard H., Wolf H., Modrow S., J. Gen. Virol., 75, 1469–1474 (1994).
- 43) Cannon P. M., Matthews S., Clark N., Byles E. D., Iourin O., Hockley D. J., Kingsman S. M., Kingsman A. J., J. Virol., 71, 3474–3483 (1997).
- 44) Morikawa Y., Kishi T., Zhang W. H., Nermut M. V., Hockley D. J., Jones I. M., J. Virol., 69, 4519–4523 (1995).
- 45) Suzuki T., Futaki S., Niwa M., Tanaka S., Ueda K., Sugiura Y., J. Biol. Chem., 277, 2437–2443 (2002).
- 46) Tang C., Loeliger E., Kinde I., Kyere S., Mayo K., Barklis E., Sun Y. N., Huang M. J., Summers M. F., J. Mol. Biol., 327, 1013–1020 (2003).
- 47) Kelly B. N., Kyere S., Kinde I., Tang C., Howard B. R., Robinson H., Sundquist W. I., Summers M. F., Hill C. P., J. Mol. Biol., 373, 355–366 (2007).
- 48) Blair W. S., Pickford C., Irving S. L., Brown D. G., Anderson M., Bazin R., Cao J. A., Ciaramella G., Isaacson J., Jackson L., Hunt R., Kjerrstrom A., Nieman J. A., Patick A. K., Perros M., Scott A. D., Whitby K., Wu H., Butler S. L., PLoS Pathog., 6, e1001220 (2010).
- 49) Kortagere S., Madani N., Mankowski M. K., et al., J. Virol., 86, 8472–8481 (2012).
- 50) Goudreau N., Lemke C. T., Faucher A.-M., Grand-Maitre C., Goulet S., Lacoste J.-E., Rancourt J., Malenfant E., Mercier J.-F., Titolo S., Mason S. W., ACS Chem. Biol., 8, 1074–1082 (2013).
- 51) Lamorte L., Titolo S., Lemke C. T., Goudreau N., Mercier J.-F., Wardrop E., Shah V. B., von Schwedler U. K., Langelier C., Banik S. S. R., Aiken C., Sundquist W. I., Mason S. W., Antimicrob. Agents Chemother., 57, 4622–4631 (2013).
- 52) Thenin-Houssier S., de Vera I. M. S., Pedro-Rosa L., Brady A., Richard A., Konnick B., Opp S., Buffone C., Fuhrmann J., Kota S., Billack B., Pietka-Ottlik M., Tellinghuisen T., Choe H., Spicer T., Scampavia L., Diaz-Griffero F., Kojetin D. J., Valente S. T., Antimicrob. Agents Chemother., 60, 2195–2208 (2016).
- 53) Thenin-Houssier S., Valente S. T., Curr. HIV Res., 14, 270–282 (2016).
- 54) Rankovic S., Ramalho R., Aiken C., Rousso I., J. Virol., 92, e00845-18 (2018).
- 55) Wu G., Zalloum W. A., Meuser M. E., Jing L., Kang D., Chen C.-H., Tian Y., Zhang F., Cocklin S., Lee K.-H., Liu X., Zhan P., Eur. J. Med. Chem., 158, 478–492 (2018).
- 56) Jiang X., Wu G., Zalloum W. A., Meuser M. E., Dick A., Sun L., Chen C.-H., Kang D., Jing L., Jia R., Cocklin S., Lee K.-H., Liu X., Zhan P., RSC Adv., 9, 28961–28986 (2019).
- 57) Singh K., Gallazzi F., Hill K. J., Burke D. H., Lange M. J., Quinn T. P., Neogi U., Sonnerborg A., Front. Microbiol., 10, 1227 (2019).
- 58) Yant S. R., Mulato A., Hansen D., et al., Nat. Med., 25, 1377–1384 (2019).
- 59) Link J. O., Rhee M. S., Tse W. C., et al., Nature (London), 584, 614–618 (2020).
- 60) Sun L., Dick A., Meuser M. E., Huang T. G., Zalloum W. A., Chen C.-H., Cherukupalli S., Xu S., Ding X., Gao P., Kang D., De Clercq E., Pannecouque C., Cocklin S., Lee K.-H., Liu X., Zhan P., J. Med. Chem., 63, 4790–4810 (2020).
- 61) Sahani R. L., Diana-Rivero R., Vernekar S. K. V., Wang L., Du H., Zhang H., Castaner A. E., Casey M. C., Kirby K. A., Tedbury P. R., Xie J., Sarafianos S. G., Wang Z., Viruses, 13, 479 (2021).
- 62) Zhang X., Sun L., Meuser M. E., Zalloum W. A., Xu S., Huang T., Cherukupalli S., Jiang X., Ding X., Tao Y., Kang D., De Clercq E., Pannecouque C., Dick A., Cocklin S., Liu X., Zhan P., Eur. J. Med. Chem., 226, 113848 (2021).
- 63) Chia T., Nakamura T., Amano M., Takamune N., Matsuoka M., Nakata H., Antimicrob. Agents Chemother., 65, e0103921 (2021).
- 64) Zhao G., Perilla J. R., Yufenyuy E. L., Meng X., Chen B., Ning J., Ahn J., Gronenborn A. M., Schulten K., Aiken C., Zhang P., Nature (London), 497, 643–646 (2013).
- 65) von Schwedler U. K., Stray K. M., Garrus J. E., Sundquist W. I., J. Virol., 77, 5439–5450 (2003).
- 66) Triad National Security, LLC. “HIV Sequence Database.”: ‹https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html›, cited 24 August, 2023.
- 67) Kobayakawa T., Yokoyama M., Tsuji K., Fujino M., Kurakami M., Boku S., Nakayama M., Kaneko M., Ohashi N., Kotani O., Murakami T., Sato H., Tamamura H., Biomolecules, 11, 208 (2021).
- 68) Wildman S. A., Crippen G. M., J. Chem. Inf. Comput. Sci., 39, 868–873 (1999).
- 69) Kobayakawa T., Yokoyama M., Tsuji K., Fujino M., Kurakami M., Onishi T., Boku S., Ishii T., Miura Y., Shinohara K., Kishihara Y., Ohashi N., Kotani O., Murakami T., Sato H., Tamamura H., RSC Adv., 13, 2156–2167 (2023).
- 70) Corey E. J., Bakshi R. K., Shibata S., J. Am. Chem. Soc., 109, 5551–5553 (1987).
- 71) Yamaguchi S., Kabuto K., Bull. Chem. Soc. Jpn., 50, 3033–3038 (1977).
- 72) Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P., “Molecular Biology of THE CELL,” 6th ed., Garland Science, New York, 2017.
- 73) Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2022.
- 74) Kobayakawa T., Amano M., Nakayama M., Tsuji K., Ishii T., Miura Y., Shinohara K., Yamamoto K., Matsuoka M., Tamamura H., RSC. Med. Chem., 14, 1973–1980 (2023).
- 75) Harada S., Koyanagi Y., Yamamoto N., Science, 229, 563–566 (1985).