Experimental
CalculationsAll calculations were performed using the Schrödinger Suite (Schrödinger Release 2023-1).
Ligand PreparationAll the calculated compounds were prepared using the LigPrep module of the Schrödinger Suite with the OPLS4 force field. The ionization state was set for pH = 7.0 ± 2.0 by using Epik. Geometry optimization and conformational searches of these compounds were performed using the MacroModel program. An OPLS4 force field was used. The calculations were performed without a solvent. A conformational search method was used for torsional sampling (Monte-Carlo Multiple Minimum, or MCMM).
Prediction of Lipophilicity and Water SolubilityThe lipophilicity and water solubility of the energetically most stable conformer were predicted using the QikProp module of the Schrödinger Suite.
Turbidity AssayNephelometric turbidity units (NTUs) were measured using a digital turbidimeter TBD700 (AS ONE, Osaka, Japan) equipped with an infrared radiation diode (wavelength: 850 nm). DMSO solutions containing each compound were diluted 1 : 100 in phosphate-buffered saline (PBS) (pH 7.4). After mixing, the measurements were conducted four times and the average NTU values were calculated.
Cell Growth AssayGrowth-suppressive effects of curcumin analogs against human colon cancer HCT116 cells were measured for 72 h. The percentage cell growth of the control, which was treated with 1% DMSO alone, was calculated and plotted, and the IC50 value was then determined. Cell viability was assayed as previously described.18,23) The data were obtained from three independent experiments.
General Information for Chemical Synthesis1H-NMR spectra were recorded with tetramethylsilane (δH 0.00), CHCl3 (δH 7.26), or CH3OH (δH 3.31) as an internal standard. Coupling constants (J) are reported in hertz (Hz). Abbreviations of multiplicity are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. Data are presented as follows: chemical shift, multiplicity, coupling constants, and integration. 13C-NMR spectra were recorded with CDCl3 (δC 77.0) and CD3OD (δC 49.0) as an internal standard. IR spectra were recorded on an FT-IR spectrophotometer and absorbance bands are reported in wavenumber (cm−1).
Column chromatography was carried out on silica gel 60 N (63–210 or 40–50 µm). Analytical TLC was carried out with 0.25 mm silica gel plates. Visualization was accomplished with ultraviolet light and anisaldehyde or phosphomolybdic acid stain, followed by heating. Reagents and solvents were purified by standard means or used as received unless otherwise noted. Dehydrated dichloromethane (CH2Cl2) and tetrahydrofuran (THF, stabilizer free) were purchased. All reactions were conducted under an argon atmosphere unless otherwise noted.
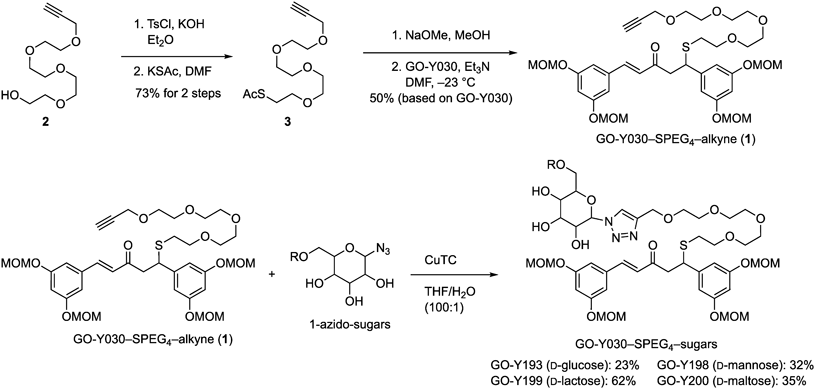
Chemical Synthesis and Compound DataAcetyl 3,6,9,12-tetraoxapentadec-14-yn-1-yl sulfide (3): KOH (2.85 g, 50.1 mmol) was added to an ice-cooled (0 °C) solution of 3,6,9,12-tetraoxapentadec-14-yn-1-ol (2, 3.87 g, 16.7 mmol) in Et2O (25 mL) After 10 min of stirring at 0 °C, TsCl (3.82 g, 20.0 mmol) was added, and the resulting mixture was stirred at room temperature for 50 min. The reaction was quenched with half-saturated aqueous NH4Cl (50 mL), and the mixture was extracted with AcOEt (160 and 80 mL). The combined organic extracts were washed with brine (80 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (6.67 g), which was used without further purification.
KSAc (2.30 g, 20.0 mmol) was added to an ice-cooled (0 °C) solution of the crude tosylate in N,N-dimethylformamide (DMF) (20 mL). After 2.5 h of stirring at room temperature, the reaction was quenched with saturated aqueous NaHCO3 (50 mL). The resulting mixture was filtered through cotton, and the filtrate was extracted with Et2O (4 × 100 mL). The combined organic extracts were washed with brine (100 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (10.7 g), which was purified by flash column chromatography (silica gel 50 g, n-hexane/AcOEt 3 : 1→AcOEt) to obtain sulfide 3 (3.54 g, 73% for two steps) as an orange oil. Rf 0.63 (AcOEt); IR (neat) 3259, 2869, 2113, 1692, 1456, 1353, 1292, 1249, 1105, 1034, 954 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 2.34 (s, 3H), 2.43 (t, J = 2.3 Hz, 1H), 3.09 (t, J = 6.5 Hz, 2H), 3.60 (t, J = 6.5 Hz, 2H), 3.63–3.72 (m, 12H), 4.21 (d, J = 2.3 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ: 28.8, 30.5, 58.4, 69.1, 69.7, 70.3, 70.4, 70.5, 70.6, 74.4, 74.5, 79.6, 195.5; HRMS (FAB) m/z [M + H]+ Calcd for C13H23O5S 291.1261. Found 291.1238.
GO-Y030-SPEG4-alkyne (1): NaOMe (347 mg, 6.42 mmol) was added to an ice-cooled (0 °C) solution of sulfide 3 (620 mg, 2.13 mmol) in MeOH (10 mL). After 1.5 h of stirring, the reaction mixture was neutralized with the Dowex 50 W × 8 acidic resin. In vacuo filtration and evaporation furnished the crude product (734 mg), which was used without further purification.
A solution of crude thiol in DMF (12 plus 2 mL rinse) was added to a cooled (−23 °C) solution of GO-Y030 (1.02 g, 2.14 mmol) and Et3N (0.35 mL, 2.52 mmol) in DMF (6.0 mL). After 2 h of stirring at −23 °C, H2O (50 mL) was added and the resulting mixture was extracted with Et2O (3 × 100 mL). The combined organic extracts were washed with brine (150 mL) and dried over anhydrous Na2SO4. In vacuo filtration and evaporation furnished the crude product (1.89 g), which was purified by flash column chromatography (silica gel 50 g, n-hexane/AcOEt 1 : 1) to give sulfide GO-Y030-SPEG4-alkyne (1, 776 mg, 50%) as a pale-yellow oil. Rf 0.15 (n-Hexane/AcOEt 1 : 1); IR (neat) 3279, 2901, 2827, 2114, 1688, 1663, 1592, 1454, 1401, 1332, 1281, 1248, 1215, 1146, 1185, 1033, 965 cm−1; 1H-NMR (400 MHz, acetone-d6) δ: 2.56 (t, J = 6.5 Hz, 2H), 2.92 (t, J = 2.5 Hz, 1H), 3.22 (dd, J = 7.3, 16.4 Hz, 1H), 3.30 (dd, J = 7.3, 16.4 Hz, 1H), 3.41 (s, 6H), 3.43 (s, 6H), 3.50–3.61 (m, 14H), 4.54 (d, J = 2.5 Hz, 2H), 4.53 (t, J = 7.3 Hz, 1H), 5.16 (s, 4H), 5.22 (s, 4H), 6.59 (t, J = 2.2 Hz, 1H), 6.76 (t, J = 2.3 Hz, 1H), 6.78 (d, J = 2.3 Hz, 2H), 6.82 (d, J = 15.9 Hz, 1H), 7.00 (d, J = 2.3 Hz, 2H), 7.57 (d, J = 15.9 Hz, 1H); 13C-NMR (150 MHz, acetone-d6) δ: 31.3, 45.6, 47.5, 56.1, 56.2, 58.5, 69.8, 70.9, 71.0, 71.2, 71.6, 75.7, 81.0, 95.1, 95.2, 104.1, 107.7, 110.20, 110.23, 127.8, 137.7, 143.1, 145.7, 159.3, 159.6, 197.0; HRMS (FAB) m/z [M + H]+ Calcd for C36H51O13S 723.3045. Found 723.3062.
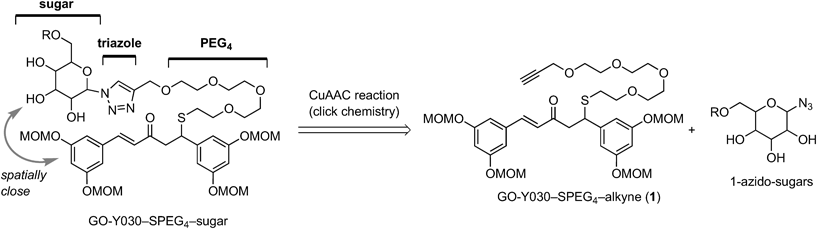
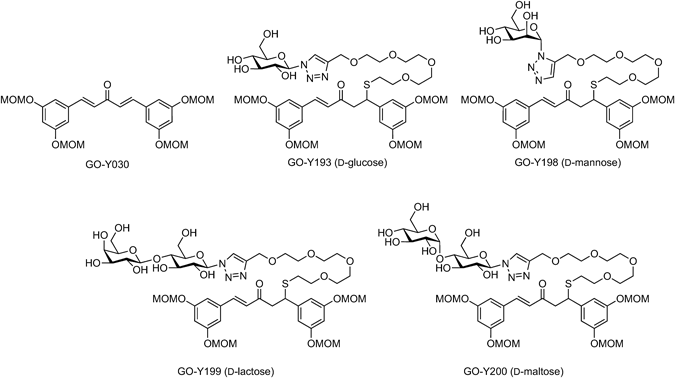
Typical Procedure for CuAAC ReactionGO-Y193: CuTC (4.1 mg, 22 µmol) was added to a mixture of GO-Y030-SPEG4-alkyne (1, 214 mg, 296 µmol) and β-D-glucopyranosyl azide24) (358 mg, 1.75 mmol) in aqueous THF (8.1 mL, THF : H2O = 100 : 1). After 2 h of stirring, the reaction mixture was concentrated in vacuo, and the residue was purified by flash column chromatography (silica gel 2.4 g, CHCl3/MeOH 19 : 1→9 : 1) to give GO-Y193 (62.0 mg, 23%) as a colorless amorphous. Rf 0.67 (CHCl3/MeOH 4 : 1); IR (neat) 3398, 1688, 1660, 1593, 1455, 1401, 1281, 1215, 1146, 1085, 1033, 924 cm−1; 1H-NMR (600 MHz, CD3OD) δ: 2.55 (t, J = 6.5 Hz, 2H), 3.18 (dd, J = 7.0, 16.1 Hz, 1H), 3.24 (dd, J = 7.9, 16.1 Hz, 1H), 3.41 (s, 6H), 3.44 (s, 6H), 3.50–3.64 (m, 17H), 3.72 (dd, J = 5.5, 12.2 Hz, 1H), 3.88 (dd, J = 2.0, 12.2 Hz, 1H), 3.90 (t, J = 9.0 Hz, 1H), 4.47 (dd, J = 7.0, 7.9 Hz, 1H), 4.62 (s, 2H), 5.12 (d, J = 2.2 Hz, 2H), 5.13 (d, J = 2.2 Hz, 2H), 5.18 (s, 4H), 5.60 (d, J = 9.1 Hz, 1H), 6.59 (t, J = 2.2 Hz, 1H), 6.75 (d, J = 16.1 Hz, 1H), 6.76 (t, J = 2.2 Hz, 1H), 6.76 (d, J = 2.2 Hz, 2H), 6.92 (d, J = 2.2 Hz, 2H), 7.48 (d, J = 16.1 Hz, 1H), 8.15 (s, 1H); 13C-NMR (150 MHz, CD3OD) δ: 31.6 (CH2), 46.4 (CH), 48.0 (CH2), 56.3 (CH3), 56.4 (CH3), 62.4 (CH2), 64.9 (CH2), 70.8 (CH2), 70.9 (CH), 71.3 (CH2), 71.51 (CH2), 71.53 (CH2), 71.55 (CH2), 72.0 (CH2), 74.0 (CH), 78.5 (CH), 81.1 (CH), 89.5 (CH), 95.5 (CH2), 95.6 (CH2), 104.7 (CH), 108.2 (CH), 110.6 (CH), 110.7 (CH), 124.3 (CH), 127.8 (CH), 137.9 (C), 144.6 (CH), 145.8 (C), 146.1 (C), 159.7 (C), 160.0 (C), 199.5 (C); HRMS (FAB) m/z [M + H]+ Calcd for C42H62N3O18S 928.37442. Found 928.3762.
GO-Y198: The reaction was performed according to the typical procedure (7.0 mL THF, 70 µL H2O, r.t., 2.5 h) employing α-D-mannopyranosyl azide25) (127 mg, 340 µmol), alkyne 1 (247 mg, 342 µmol), and CuTC (5.2 mg, 27.3 µmol). GO-Y198 (102 mg, 32%) was obtained as a colorless amorphous solid from the crude product after flash column chromatography (silica gel 2.3 g, CHCl3/MeOH 19 : 1→9 : 1). Rf 0.13 (AcOEt/MeOH 9 : 1); IR (neat) 3399, 2903, 1685, 1654, 1593, 1456, 1400, 1332, 1281, 1215, 1146, 1084, 1033, 924 cm−1; 1H-NMR (600 MHz, CD3OD) δ: 2.53 (t, J = 6.5 Hz, 2H), 3.16 (dd, J = 7.0, 15.8 Hz, 1H), 3.20 (dd, J = 7.9, 15.8 Hz, 1H), 3.39 (s, 6H), 3.43 (s, 6H), 3.49–3.52 (m, 4H), 3.55–3.63 (m, 11H), 3.72–3.82 (m, 3H), 4.07 (dd, J = 3.3, 8.7 Hz, 1H), 4.45 (dd, J = 6.9, 7.9 Hz, 1H), 4.68 (t, J = 3.3 Hz, 1H), 4.80 (s, 2H), 5.11 (d, J = 6.8 Hz, 2H), 5.14 (d, J = 6.8 Hz, 2H), 5.16 (s, 4H), 6.02 (d, J = 2.6 Hz, 1H), 6.57 (t, J = 2.1 Hz, 1H), 6.72 (d, J = 16.1 Hz, 1H), 6.74 (t, J = 2.1 Hz, 1H), 6.74 (d, J = 2.0 Hz, 2H), 6.90 (d, J = 2.0 Hz, 2H), 7.46 (d, J = 16.1 Hz, 1H), 8.12 (s, 1H); 13C-NMR (150 MHz, CD3OD) δ: 31.6 (CH2), 46.3 (CH), 48.0 (CH2), 56.37 (CH3), 56.42 (CH3), 62.5 (CH2), 64.9 (CH2), 68.5 (CH), 70.1 (CH), 70.8 (CH2), 71.2 (CH2), 71.45 (CH2), 71.48 (CH2), 71.51 (CH2), 71.9 (CH2), 72.5 (CH), 78.5 (CH), 88.3 (CH), 95.5 (CH2), 95.6 (CH2), 104.7 (CH), 108.2 (CH), 110.6 (CH), 110.7 (CH), 125.0 (CH), 127.8 (CH), 137.9 (C), 144.5 (CH), 145.8 (C), 146.3 (C), 159.7 (C), 159.9 (C), 199.3 (C); HRMS (FAB) m/z [M + H]+ Calcd for C42H62N3O18S 928.3744. Found 928.3752.
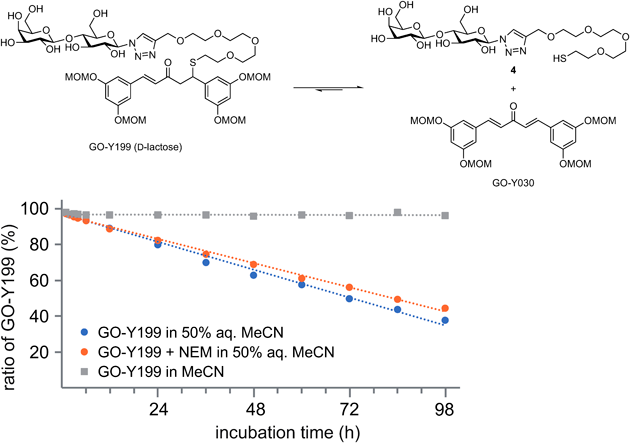
GO-Y199: The reaction was performed according to the typical procedure (6.0 mL THF, 120 µL H2O, r.t., 2 h) employing β-D-lactosyl azide24) (109 mg, 296 µmol), alkyne 1 (214 mg, 296 µmol), and CuTC (6.2 mg, 32.5 µmol). GO-Y199 (199 mg, 62%) was obtained as a colorless amorphous solid from the crude product after flash column chromatography (twice, silica gel 3 g, CHCl3/MeOH 9 : 1→4 : 1, silica gel 2.3 g, CHCl3/MeOH 9 : 1→17 : 3). Rf 0.20 (CHCl3/MeOH 4 : 1); IR (neat) 3389, 1660, 1593, 1455, 1440, 1401, 1333, 1281, 1241, 1215, 1146, 1084, 1034, 924 cm−1; 1H-NMR (600 MHz, CD3OD) δ: 2.55 (t, J = 6.6 Hz, 2H), 3.18 (dd, J = 7.1, 15.9 Hz, 1H), 3.23 (dd, J = 7.9, 15.9 Hz, 1H), 3.41 (s, 6H), 3.45 (s, 6H), 3.47–3.64 (m, 17H), 3.74–3.83 (m, 6H), 3.89 (m, 2H), 3.97 (t, J = 9.1 Hz, 1H), 4.41 (d, J = 7.8 Hz, 1H), 4.47 (dd J = 6.6, 7.9 Hz, 1H), 4.62 (s, 2H), 5.12 (d, J = 6.9 Hz, 2H), 5.14 (d, J = 6.9 Hz, 2H), 5.18 (s, 4H), 5.63 (d, J = 9.1 Hz, 1H), 6.58 (t, J = 2.2 Hz, 1H), 6.74 (d, J = 16.2 Hz, 1H), 6.76 (t, J = 2.1 Hz, 1H), 6.76 (d, J = 2.2 Hz, 2H), 6.92 (d, J = 2.1 Hz, 2H), 7.48 (d, J = 16.2 Hz, 1H), 8.16 (s, 1H); 13C-NMR (150 MHz, CD3OD) δ: 31.6 (CH2), 46.4 (CH), 48.1 (CH2), 56.3 (CH3), 56.4 (CH3), 61.6 (CH2), 62.5 (CH2), 65.0 (CH2), 70.3 (CH), 70.8 (CH2), 71.3 (CH2), 71.51 (CH2), 71.56 (CH2), 71.57 (CH2), 71.59 (CH2), 72.0 (CH2), 72.5 (CH), 73.7 (CH), 74.9 (CH), 76.9 (CH), 77.1 (CH), 79.6 (CH), 79.8 (CH), 89.3 (CH), 95.56 (CH2), 95.62 (CH2), 104.8 (CH), 105.1 (CH), 108.2 (CH), 110.65 (CH), 110.72 (CH), 124.3 (CH), 127.8 (CH), 137.9 (C), 144.6 (CH), 145.8 (C), 146.1 (C), 159.8 (C), 160.0 (C), 199.6 (C); HRMS (FAB) m/z [M + H]+ Calcd for C48H72N3O23S 1090.4272. Found 1090.4265.
GO-Y200: The reaction was performed according to the typical procedure (10 mL THF, 100 µL H2O, r.t., 2 h) employing β-D-maltosyl azide24) (160 mg, 436 µmol), alkyne 1 (226 mg, 313 µmol), and CuTC (3.3 mg, 17.3 µmol). GO-Y200 (133 mg, 35%) was obtained as a colorless amorphous solid from the crude product after flash column chromatography (silica gel 5.5 g, CHCl3/MeOH 9 : 1→17 : 3). Rf 0.08 (CHCl3/MeOH 8 : 1); IR (neat) 3386, 2904, 1684, 1661, 1594, 1541, 1506, 1456, 1400, 1334, 1281, 1216, 1146, 1084, 1033, 968 cm−1; 1H-NMR (600 MHz, CD3OD) δ: 2.56 (t, J = 6.5 Hz, 2H), 3.18 (dd, J = 6.9, 16.0 Hz, 1H), 3.24 (dd, J = 7.9, 16.0 Hz, 1H), 3.42 (s, 6H), 3.46 (s, 6H), 3.48 (m, 1H), 3.53–3.56 (m, 4H), 3.59–3.70 (m, 15H), 3.76 (t, J = 9.2 Hz, 1H), 3.82–3.90 (m, 4H), 3.96 (t, J = 9.2 Hz, 1H), 4.48 (dd J = 6.9, 7.9 Hz, 1H), 4.63 (s, 2H), 5.13 (d, J = 6.8 Hz, 2H), 5.15 (d, J = 6.8 Hz, 2H), 5.19 (s, 4H), 5.24 (d, J = 3.9 Hz, 1H), 5.63 (d, J = 9.2 Hz, 1H), 6.59 (t, J = 2.2 Hz, 1H), 6.75 (d, J = 16.3 Hz, 1H), 6.77 (t, J = 2.2 Hz, 1H), 6.77 (d, J = 2.2 Hz, 2H), 6.94 (d, J = 2.2 Hz, 2H), 7.49 (d, J = 16.3 Hz, 1H), 8.17 (s, 1H); 13C-NMR (150 MHz, CD3OD) δ: 31.6 (CH2), 46.4 (CH), 48.1 (CH2), 56.3 (CH3), 56.4 (CH3), 61.9 (CH2), 62.8 (CH2), 65.0 (CH2), 70.8 (CH), 71.3 (CH2), 71.53 (CH), 71.57 (CH2), 71.58 (CH2), 71.60 (CH2), 72.1 (CH2), 73.6 (CH), 74.2 (CH), 74.9 (CH), 75.1 (CH), 78.2 (CH), 79.7 (CH), 80.4 (CH), 89.4 (CH), 95.57 (CH2), 95.63 (CH2), 103.0 (CH), 104.8 (CH), 108.2 (CH), 110.66 (CH), 110.72 (CH), 124.3 (CH), 127.8 (CH), 138.0 (C), 144.6 (CH), 145.8 (C), 146.1 (C), 159.8 (C), 160.0 (C), 199.6 (C); HRMS (FAB) m/z [M + H]+ Calcd for C48H72N3O23S 1090.4272. Found 1090.4303.