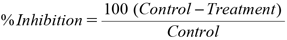Abstract
The development of polymeric nanoparticles (NPs) from preformed polymers usually requires the use of organic solvents and is more expensive. Hence, in this work, the development of polymeric nanoparticles by in situ aqueous dispersion polymerization from the monomers was set as an objective. Acrylonitrile monomer based polymeric NPs comprising Lamivudine (LMV) as a model drug were prepared using the aqueous dispersion polymerization technique. A quality by design approach was applied to optimise various formulation and process factors viz. monomer concentration, initiator concentration, stabilizer concentration and polymerization temperature. Polymerization time (PT), entrapment efficiency (EE), particle size (PS), and drug release rate constant (k) were taken as the responses to define the quality of the prepared NPs. Design of experiments analysis followed by optimization was performed to identify the optimized combination of the factors. Later, the optimized formulation was studied for the physical state of the LMV in the nanoparticles, surface morphology of the NPs and cytotoxicity studies. The optimized formulation was found to have 91.7 min. of PT, 81.4% of EE, 253 nm of PS and a k value of 0.262 h−1 (18 h to release 99%). The cytotoxicity studies indicated that the NPs were highly safe to use. These results altogether inferred that LMV contained NPs were developed effectively from the acrylonitrile monomer by the aqueous dispersion polymerization method.
Introduction
Synthetic preformed polymers have been playing a vital role in the development of drug delivery systems (DDS), especially in controlled and targeted DDS. These polymers are commonly used in the development of DDS, such as matrix tablets, reservoir tablets, microparticles, nanoparticles, transdermal systems, implants, etc., for the purpose of extended release or targeted release or both.1,2) Considering the advancements in technology, present- day drug development researchers are moving towards the development of multiarticulate DDS from single-unit systems. The multiparticulate systems are capable of improving the stability and bioavailability of the drugs even through oral route; also, they are capable of achieving both controlled as well as targeted drug release.3) Among various multiarticulate systems, polymeric nanoparticles (NPs) stand first owing to their unique advantages like suitability to any type of final dosage form (as they can be made into tablets, capsules, oral liquid, parenteral liquids and also for transdermal systems), high physical stability even in oral route also besides providing modified and targeted delivery of drugs. Several synthetic polymers like ethyl cellulose, polymethacrylic acids, polyvinyl acetate, polyvinyl alcohol, polylactic acid, polygylocolic acid, polyacrylonitrile etc. have been widely explored for the development of polymeric nanoparticles for various drug specific objectives. Several technologies based on precipitation, coacervation and solvent evaporation have been employed in developing polymeric NPs.4–6)
Almost all of the techniques employed in the development of various types of DDS use preformed polymers only, but these preformed polymers suffer two major drawbacks. These synthetic polymers, except a very few which are water soluble, require organic solvents during their development into several DDS, and hence, strict environmental safety regulations have to be adopted during their largescale manufacturing.4,7) Further, polymers with any unique characteristics like biocompatibility, biodegradability, pH-sensitivity, high thermal of chemical stability are more expensive These setbacks always demand an alternative to make the development of DDS simpler and more economical yet effective.
In this context, single-step aqueous dispersion polymerization (ADP), an in-situ polymerization technique with monomers for the development of NPs is one of the best alternatives to the use of preformed polymers.8) In this ADP technique, monomer, polymerization initiator and stabilizer are solubilized in aqueous solution of a drug. Upon continuous mixing under suitable temperature, polymerization is started and the in-situ formed polymer with entrapped drug gets precipitated from the aqueous solution due to its insolubility in water.9) This technique is a single-step process and does not require organic solvents or any sophisticated homogenization equipment. Simple mixing with heat provision is sufficient to prepare the NPs. However, several formulation factors like concentrations of monomer, initiator and stabilizer; and process factors like mixing speed and temperature influence the polymerization time and characteristics of the NPs. Upon thorough experimental investigation, reports by few authors on this technique for developing polymeric nanoparticles were observed.10–12) Polyacrylonitrile (PAN) is one such polymer which can be prepared by ADP technique. Due to its excellent thermal and mechanical stability as well as the biocompatibility and non-toxic nature,13) PAN is commonly used in biomedical applications.14) Except a few reports by Lian et al.15) and Lv et al.16) no significant reports were found in exploring this technique for achieving controlling release of the incorporated drug. Hence, considering the scope of this technique, in the current research work presented in this study, the ADP technique was explored to develop PAN nanoparticles for lamivudine (LMV), which was taken as a model drug. Application of PAN in the development of DDS is growing owing to its high chemical stability and excellent barrier properties. PAN is water insoluble and soluble in organic solvents. Hence the preformed polymer requires use of organic solvents for the development of NPs. Whereas acrylonitrile monomer is high polar and water soluble. Hence, ADP technique is suitable for the development of PAN NPs. So, the development of LMV containing PAN NPs (LPANs) were aimed to be developed by the ADP technique in this work. Besides, development of LPANs with considerably high drug entrapment and more controlled release of even a high water-soluble drug like LMV were further taken as the primary objectives of this work. Various formulation and process variables of the ADP technique may influence the characteristics of the LPANs along with the polymerization time.12) Hence this work was carried out in analyzing the influences of four different formulation/process variables (factors) on four different characteristics of the LPANs (responses) followed by their optimization. For this purpose, statistical quality by design (QbD) approach17) was adopted using Design Expert software in this work.
Experimental
MaterialsLamivudine was obtained as a gift sample from Hetero Drugs Pvt. Ltd., Hyderabad, India. Acrylonitrile and Potassium persulphate were gifted samples from Loba Chemie Pvt. Ltd., Mumbai, India. Sodium lauryl sulphate and Hydrochloric acid were procured from Thermo
Fischer Scientific India Pvt. Ltd., Mumbai, India. All other solvents used are of analytical grade.
Development of LMV-Polyacrylonitrile Nanoparticles (LPANs)LPANs development was carried out using dispersion polymerization technique with an object of achieving nanoparticles in less possible preparation time yet having high drug entrapment less size with controlled drug release. Box–Behnken Design (BBD)18,19) under QbD approach with help of Stat Ease Design Expert v13.0 was applied as a tool to understand the relation between different independent factors and quality characteristics of the nanoparticles. From the past experiences and literature search, Factor A: Concentration of monomer (Acrylonitrile); Factor B: Concentration of initiator (Potassium persulphate); Factor C: Concentration of stabilizer (Sodium lauryl sulphate); and Factor D: Reaction temperature were taken as the independent factors. On the other side, R1: Polymerization time (PT); R2: Entrapment efficiency (EE); R3: Particle size (PS); and R4: Drug release rate constant (k) were taken as the responses which represent the quality of the process and the product obtained. The BBD with three blocks and two center points per block was taken so as to minimize the error and, the suggested combinations of the factors are displayed in Table 1.
Table 1. Formulation Combinations of Lamivudine Polymeric Nano Particles
| S. No. | Formulation code | Factor A (conc. of monomer) % (w/v) | Factor B (conc. of Initiator) % (w/v) | Factor C (conc. of stabilizer) % (w/v) | Factor D (polymerization temp.) |
|---|
| 1. | FLPAN1.1 | 5 | 0.3 | 1 | 65 |
| 2. | FLPAN1.2 | 5 | 0.7 | 1 | 65 |
| 3. | FLPAN1.3 | 7 | 0.5 | 0.5 | 60 |
| 4. | FLPAN1.4 | 7 | 0.5 | 0.5 | 70 |
| 5. | FLPAN1.5 | 7 | 0.5 | 1 | 65 |
| 6. | FLPAN1.6 | 7 | 0.5 | 1 | 65 |
| 7. | FLPAN1.7 | 7 | 0.5 | 1.5 | 60 |
| 8. | FLPAN1.8 | 7 | 0.5 | 1.5 | 70 |
| 9. | FLPAN1.9 | 9 | 0.3 | 1 | 65 |
| 10. | FLPAN1.10 | 9 | 0.7 | 1 | 65 |
| 11. | FLPAN2.1 | 5 | 0.5 | 1 | 60 |
| 12. | FLPAN2.2 | 5 | 0.5 | 1 | 70 |
| 13. | FLPAN2.3 | 7 | 0.3 | 0.5 | 65 |
| 14. | FLPAN2.4 | 7 | 0.3 | 1.5 | 65 |
| 15. | FLPAN2.5 | 7 | 0.5 | 1 | 65 |
| 16. | FLPAN2.6 | 7 | 0.5 | 1 | 65 |
| 17. | FLPAN2.7 | 7 | 0.7 | 0.5 | 65 |
| 18. | FLPAN2.8 | 7 | 0.7 | 1.5 | 65 |
| 19. | FLPAN2.9 | 9 | 0.5 | 1 | 60 |
| 20. | FLPAN2.10 | 9 | 0.5 | 1 | 70 |
| 21. | FLPAN3.1 | 5 | 0.5 | 0.5 | 65 |
| 22. | FLPAN3.2 | 5 | 0.5 | 1.5 | 65 |
| 23. | FLPAN3.3 | 7 | 0.3 | 1 | 60 |
| 24. | FLPAN3.4 | 7 | 0.3 | 1 | 70 |
| 25. | FLPAN3.5 | 7 | 0.5 | 1 | 65 |
| 26. | FLPAN3.6 | 7 | 0.5 | 1 | 65 |
| 27. | FLPAN3.7 | 7 | 0.7 | 1 | 60 |
| 28. | FLPAN3.8 | 7 | 0.7 | 1 | 70 |
| 29. | FLPAN3.9 | 9 | 0.5 | 0.5 | 65 |
| 30. | FLPAN3.10 | 9 | 0.5 | 1.5 | 65 |
LPANs were developed using dispersion polymerization technique.20–22) Briefly, 1 g of LMV was first dissolved in sufficient volume of double distilled water. To this drug solution, acrylonitrile, potassium persulphate and Sodium lauryl sulphate (SLS) were added and mixed to dissolve and the volume of this solution was made to 20 mL with double distilled water. This solution was kept for stirring at 100 min−1 (revolutions per minute) on at a specified temperature. The quantities of the materials (other than LMV) and the reaction temperature were taken according to the BBD as mentioned in the Table 1. The initiation of the polymerization reaction was indicated by conversion of the clear solution into dispersion with formation of precipitate. The completion of the reaction was identified by the formation of white precipitate at the bottom with clear/transparent nature of the remaining medium. The obtained LPANs were collected using centrifugation followed by multiple washing cycles.
Evaluation of NanoparticlesEntrapment EfficiencyEntrapment efficiency was determined using centrifugation method. Briefly the colloidal suspension of polymeric nanoparticles was centrifuged for 30 min at 8000 min−1 at 4 °C. The supernatant was collected and estimated for its unentrapped drug content23) which was substituted in the below mentioned formula to get the EE.
Particle Size AnalysisParticle size is one of the key factor in deciding of the drug release pattern and other physicochemical properties. The particle size analysis was performed for all the formulations using Malvern Zeta sizer nano series24) followed by the statistical analysis to understand the impact different factors on particle size.
In-vitro Drug Release StudyIn-vitro drug release studies were performed using the dialysis bag method25) under continuous stirring at 100 min−1. Briefly, the polymeric nanoparticles were suspended in purified water and transferred into a dialysis bag, sealed and suspended into a beaker containing 0.1N HCl in a beaker at 37 ± 0.5 °C and continuous agitation was applied at 100 min−1. Aliquots of samples were collected up to 24 h and replaced with fresh buffer. The samples were analyzed using UV visible spectrophotometer at 270 nm.
Drug Release KineticsTo understand the drug release mechanism from the polymeric nanoparticles, the drug release data was fitted into zero order, first order, Higuchi and Korsemeyer–Peppas models and the regression coefficient values were reported. The formulae for these kinetic models are
Where, Dt is the % drug released at time t; k is rate constant of the respective model; UDt is the % drug unreleased at time t; D・ is the % drug released at infinite time; and n is the Korsemeyer–Peppas coefficient.
The release rate constant in the First–order model was calculated by multiplying the slope the plot with numeric value of “ − 2.303” according on its model equation.
Design of Experiments (DoE) Validation and OptimizationDesign validation was performed for the selected BBD model using Design Expert software v13.0 to identify the significance of the formulation and process parameters on the all the four responses. Further, ANOVA was applied to the suggested model to find the suitability of the model. After successful model selection, graphical optimization was performed by using desirability functions approach to identify the best combination of the formulation factors’ levels. The desirability criteria were taken by setting constraints to the responses in accordance with the set objective. Hence, the constraints were taken as minimizing PT with an upper limit of 100 min., maximizing EE with a lower limit of 75%, minimizing PS with an upper limit of 250 nm, and minimizing k value of with an upper limit of 0.3 h−1.
Differential Scanning Calorimetry (DSC)The DSC was carried out for the prepared formulation and for the plain drug to understand the changes in melting point of the drug using DSC-60 model of Shimadzu Corporation Kyoto, Japan. Briefly, the samples were sealed hermetically onto aluminum pan and empty sealed aluminum pan was used as a reference. The samples were scanned over a temperature range of 27 to 400 °C at 10 °C/min heating rate.
Surface MorphologyThe formulated Lamivudine loaded polymeric nanoparticles were subjected for the determination of surface morphology using transmission electron microscopy (TEM) JEOL MAKE, JEM 2100 model TEM.26)
Cytotoxicity StudiesThe cell viability study was performed using 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) Assay with three independent experiments using six concentrations of the compounds in triplicates. The trypan blue assay was performed by trypnizing the cells in cell suspension to detect the cell viability. Using a haemocytometer, the cells were counted and seeded at density of 5.0 × 103 cells/well in 100 µL media in 96 well plate culture medium and incubated overnight at 37 °C. After incubation, the old media was replaced with 100 µL of fresh media with different concentrations of test compound in represented wells in 96 plates. After 48 h, the drug solution was discarded and fresh media with MTT solution (0.5 mg/mL) was added to each well and the plates were incubated at 37 °C for 3 h. At the end of incubation time, precipitates were formed because of the reduction of the MTT salt to chromophore formazan crystals by the cells having metabolically active mitochondria.27) The optical density of solubilized crystals in dimethyl sulfoxide (DMSO) was measured at 570 nm on a microplate reader. The percentage growth inhibition was calculated using the following formula.
Results and Discussion
In the current research work, the LPANs were prepared as 30 formulations as per the three block BBD by varying different formulation and process parameters by employing dispersion polymerization method. All the factors varied were taken at three different levels. In this work, LMV, the active pharmaceutical ingredient was considered as the model drug and taken at 1 g in each formulation. Completion of the polymerization reaction was identified by the development of white precipitate with clear medium. The obtained precipitate was collected by centrifugation and washed several times to remove any unreacted monomer amount. Later, it was dried and subjected to characterization testing.
Characterization of the ResponsesThe formulated LPANs were evaluated for its particles size, entrapment efficiency and the polymerization time. The polymerization time was found to be in the range of 64 min. to 850 min, whereas the entrapment efficiency is in the range of 52.9 to 75.8% and the particle size was in the range of 160.2 to 315.5 nm. These results are presented in Table 2. The results of drug release studies are illustrated in Fig. 1 and maximum control of the drug release was observed up to 24 h. And the results of drug release kinetic analysis including the release rate constant (k) are given in Table 3. These results designated that that almost all the formulations were found to follow first order kinetics of drug release. The exponent (n value) form the Korsemeyer-Peppas plots of majority of the formulations was found to be in the range of 0.45–0.89, thus indicating the drug release mechanism was non-Fickian diffusion.
Table 2. Results of Physicochemical Characterization of LPAN (Expressed in Mean ± Standard Deviation for
n = 3)
| S. No. | Formulation code | R1: Polymerization time (min) | R2: Entrapment efficiency (%) | R3: Particle size (nm) |
|---|
| 1 | FLPAN1.1 | 221 ± 28 | 55.6 ± 3.4 | 160.2 ± 10.7 |
| 2 | FLPAN1.2 | 201 ± 17 | 52.9 ± 2.7 | 188.7 ± 14.3 |
| 3 | FLPAN1.3 | 164 ± 23 | 64.6 ± 1.9 | 231.3 ± 20.6 |
| 4 | FLPAN1.4 | 130 ± 15 | 63.8 ± 2.2 | 239.6 ± 21.5 |
| 5 | FLPAN1.5 | 144 ± 13 | 65.9 ± 2.6 | 219.2 ± 19.2 |
| 6 | FLPAN1.6 | 146 ± 18 | 66.9 ± 1.8 | 217.8 ± 16.5 |
| 7 | FLPAN1.7 | 159 ± 16 | 71.6 ± 1.7 | 189.9 ± 18.1 |
| 8 | FLPAN1.8 | 124 ± 11 | 70.5 ± 3.1 | 185.1 ± 20.3 |
| 9 | FLPAN1.9 | 90 ± 10 | 75.8 ± 2.8 | 289.1 ± 26.4 |
| 10 | FLPAN1.10 | 64 ± 12 | 74.6 ± 1.4 | 315.5 ± 24.9 |
| 11 | FLPAN2.1 | 219 ± 24 | 54.2 ± 2.3 | 168.7 ± 20.6 |
| 12 | FLPAN2.2 | 175 ± 26 | 54.0 ± 2.6 | 181.3 ± 15.7 |
| 13 | FLPAN2.3 | 189 ± 19 | 72.3 ± 1.8 | 194.2 ± 16.9 |
| 14 | FLPAN2.4 | 153 ± 20 | 74.1 ± 2.5 | 168.5 ± 11.7 |
| 15 | FLPAN2.5 | 141 ± 12 | 67.5 ± 3.1 | 221.3 ± 21.5 |
| 16 | FLPAN2.6 | 138 ± 17 | 67.0 ± 2.7 | 219.7 ± 20.6 |
| 17 | FLPAN2.7 | 114 ± 13 | 56.1 ± 1.9 | 273.9 ± 24.3 |
| 18 | FLPAN2.8 | 110 ± 16 | 59.0 ± 2.4 | 237.1 ± 26.2 |
| 19 | FLPAN2.9 | 88 ± 10 | 75.3 ± 3.4 | 292.6 ± 18.7 |
| 20 | FLPAN2.10 | 67 ± 14 | 75.1 ± 1.7 | 309.4 ± 23.8 |
| 21 | FLPAN3.1 | 194 ± 25 | 53.2 ± 1.2 | 175.9 ± 13.6 |
| 22 | FLPAN3.2 | 189 ± 29 | 54.5 ± 1.6 | 138.4 ± 11.4 |
| 23 | FLPAN3.3 | 203 ± 18 | 73.7 ± 3.4 | 191.5 ± 16.9 |
| 24 | FLPAN3.4 | 162 ± 15 | 72.8 ± 2.3 | 203.4 ± 21.7 |
| 25 | FLPAN3.5 | 139 ± 11 | 68.1 ± 1.4 | 220.6 ± 19.5 |
| 26 | FLPAN3.6 | 137 ± 16 | 68.4 ± 2.6 | 214.9 ± 18.3 |
| 27 | FLPAN3.7 | 115 ± 12 | 58.3 ± 2.1 | 269.2 ± 23.1 |
| 28 | FLPAN3.8 | 98 ± 14 | 57.8 ± 2.3 | 284.2 ± 26.2 |
| 29 | FLPAN3.9 | 75 ± 10 | 74.6 ± 2.8 | 319.3 ± 20.8 |
| 30 | FLPAN3.10 | 72 ± 13 | 80.7 ± 3.4 | 273.8 ± 22.4 |
Table 3. Results of Drug Release Kinetic Studies of the LPANs Including the Results of R4
| S. No. | Formulation code | Regression values | Peppa’s n-value | R4: Drug release rate constant–k (h−1) |
|---|
| Zero order | First order | Higuchi’s |
|---|
| 1. | FLPAN1.1 | 0.556 | 0.961 | 0.952 | 0.525 | 0.5204 |
| 2. | FLPAN1.2 | 0.534 | 0.969 | 0.958 | 0.517 | 0.4536 |
| 3. | FLPAN1.3 | 0.375 | 0.921 | 0.918 | 0.503 | 0.3915 |
| 4. | FLPAN1.4 | 0.548 | 0.929 | 0.948 | 0.579 | 0.3339 |
| 5. | FLPAN1.5 | 0.469 | 0.909 | 0.933 | 0.559 | 0.3385 |
| 6. | FLPAN1.6 | 0.641 | 0.875 | 0.969 | 0.565 | 0.3569 |
| 7. | FLPAN1.7 | 0.609 | 0.968 | 0.961 | 0.569 | 0.373 |
| 8. | FLPAN1.8 | 0.549 | 0.879 | 0.921 | 0.625 | 0.3316 |
| 9. | FLPAN1.9 | 0.754 | 0.960 | 0.958 | 0.702 | 0.2556 |
| 10. | FLPAN1.10 | 0.757 | 0.968 | 0.982 | 0.691 | 0.2233 |
| 11. | FLPAN2.1 | 0.542 | 0.970 | 0.948 | 0.532 | 0.4905 |
| 12. | FLPAN2.2 | 0.640 | 0.922 | 0.970 | 0.538 | 0.4536 |
| 13. | FLPAN2.3 | 0.545 | 0.964 | 0.952 | 0.544 | 0.4053 |
| 14. | FLPAN2.4 | 0.618 | 0.963 | 0.957 | 0.572 | 0.4030 |
| 15. | FLPAN2.5 | 0.467 | 0.919 | 0.936 | 0.544 | 0.3569 |
| 16. | FLPAN2.6 | 0.501 | 0.919 | 0.940 | 0.559 | 0.3638 |
| 17. | FLPAN2.7 | 0.543 | 0.928 | 0.950 | 0.587 | 0.3062 |
| 18. | FLPAN2.8 | 0.610 | 0.944 | 0.960 | 0.614 | 0.2993 |
| 19. | FLPAN2.9 | 0.824 | 0.985 | 0.970 | 0.718 | 0.2441 |
| 20. | FLPAN2.10 | 0.832 | 0.987 | 0.978 | 0.717 | 0.2256 |
| 21. | FLPAN3.1 | 0.597 | 0.984 | 0.967 | 0.531 | 0.4652 |
| 22. | FLPAN3.2 | 0.439 | 0.927 | 0.919 | 0.521 | 0.4582 |
| 23. | FLPAN3.3 | 0.409 | 0.938 | 0.919 | 0.519 | 0.4352 |
| 24. | FLPAN3.4 | 0.453 | 0.913 | 0.929 | 0.521 | 0.3851 |
| 25. | FLPAN3.5 | 0.673 | 0.976 | 0.972 | 0.592 | 0.3661 |
| 26. | FLPAN3.6 | 0.719 | 0.961 | 0.960 | 0.562 | 0.3707 |
| 27. | FLPAN3.7 | 0.452 | 0.901 | 0.933 | 0.559 | 0.3293 |
| 28. | FLPAN3.8 | 0.487 | 0.881 | 0.929 | 0.595 | 0.2970 |
| 29. | FLPAN3.9 | 0.873 | 0.991 | 0.972 | 0.722 | 0.2326 |
| 30. | FLPAN3.10 | 0.867 | 0.990 | 0.972 | 0.746 | 0.2279 |
Under DoE analysis, all the four responses were analyzed by sequential model sum of squares from the software. The results indicated that the all the four responses were influenced by linear model with the factors. The linear regression equations for the responses in coded factors but not actual factors were as follows
These regression equations contain only main effects and did not contain interaction terms and squared terms even though the selected design was BBD. This is because the linear regression model was suggested for every response but not quadratic model from the sequential model sum of squares analysis. These regression equations are useful in understanding the magnitude of influence of every factor by observing their coefficients. These factors taken were standardized as their influences on the responses were found significant from ANOVA. Hence, the response surface linear model was tested for its significance along with the factor terms in case of all the four responses by ANOVA. The results are presented in Table 4, and these results were designating that the linear regression models in case of all the four responses were significant. The predicted vs. actual plots for all the four responses are illustrated in Fig. 2. These plots exhibited that all the data points were uniformly distributed on either side of the 45° line and thus indicating no transformation of the data was needed for statistical analysis and optimization. Thus, these results altogether signified that the data in the form of linear model could be progressed for optimization.
Table 4. ANOVA Test Results for the Linear Model of All the Responses
| Source | Sum of squares | Degrees of freedom | Mean sum of squares | F-value | p-value | Inference* |
|---|
| R1: PT |
| Block | 199.4 | 2 | 99.7 | | | |
| Model | 57687.5 | 4 | 14421.9 | 124.26 | < 0.0001 | Significant |
| A-Monomer conc. | 46004.1 | 1 | 46004.1 | 396.38 | < 0.0001 | Significant |
| B-Initiator conc. | 8321.3 | 1 | 8321.3 | 71.70 | < 0.0001 | Significant |
| C-Stabilizer conc. | 290.1 | 1 | 290.1 | 2.50 | 0.1275 | |
| D-Polymerization Temp | 3072.0 | 1 | 3072.0 | 26.47 | < 0.0001 | Significant |
| Residual | 2669.4 | 23 | 116.1 | | | |
| Cor Total | 60556.3 | 29 | | | | |
| R2: EE |
| Block | 3.80 | 2 | 1.9 | | | |
| Model | 1860.6 | 4 | 465.2 | 59.11 | < 0.0001 | Significant |
| A-Monomer conc. | 1445.4 | 1 | 1445.4 | 183.67 | < 0.0001 | Significant |
| B-Initiator conc. | 358.6 | 1 | 358.6 | 45.57 | < 0.0001 | Significant |
| C-Stabilizer conc. | 55.5 | 1 | 55.5 | 7.05 | 0.0142 | Significant |
| D-Polymerization Temp | 1.1 | 1 | 1.1 | 0.1450 | 0.7069 | |
| Residual | 181.0 | 23 | 7.9 | | | |
| Cor Total | 2045.4 | 29 | | | | |
| R3: PS |
| Block | 150.7 | 2 | 75.4 | | | |
| Model | 67604.9 | 4 | 16901.2 | 77.70 | < 0.0001 | Significant |
| A-Monomer conc. | 51548.5 | 1 | 51548.5 | 237.00 | < 0.0001 | Significant |
| B-Initiator conc. | 10902.2 | 1 | 10902.2 | 50.12 | < 0.0001 | Significant |
| C-Stabilizer conc. | 4856.2 | 1 | 4856.2 | 22.33 | < 0.0001 | Significant |
| D-Polymerization Temp | 298.0 | 1 | 298.0 | 1.37 | 0.2538 | |
| Residual | 5002.6 | 23 | 217.5 | | | |
| Cor total | 60556.3 | 29 | | | | |
| R4: k |
| Block | 0.0000 | 2 | 0.0000 | | | |
| Model | 0.1963 | 4 | 0.0491 | 299.32 | < 0.0001 | Significant |
| A-Monomer conc. | 0.1704 | 1 | 0.1704 | 1039.63 | < 0.0001 | Significant |
| B-Initiator conc. | 0.0208 | 1 | 0.0208 | 127.10 | < 0.0001 | Significant |
| C-Stabilizer conc. | 0.0002 | 1 | 0.0002 | 1.27 | 0.2712 | |
| D-Polymerization Temp | 0.0048 | 1 | 0.0048 | 29.28 | < 0.0001 | Significant |
| Residual | 0.0038 | 23 | 0.0002 | | | |
| Cor Total | 0.2001 | 29 | | | | |
* The model terms with p-value less than 0.05 are significant.
The impacts of the factors on PT are presented in Figs. 3(a) and 3(b). It was inferred that higher monomer concentration, the time taken for the completion of the polymerization reaction was lesser which might be attributed to the increase in the initial solvency of the reaction medium at high concentration of the monomer leading to the increase in the polymerization rate. Also, generation of free radical loci for reaction are more at higher monomer concentration which lead to rapid polymerization. These results were found in accordance with those reported by Dauletov et al.28) and Kongkaew et al.29) As the initiator concentration was increased, the time taken for the polymerization reaction to be completed was lesser. This could be owing to the increase in the speed of free radicles formation which thereby resulted in the rapid polymerization rate.28,30) It was inferred that in the current research work, the effect of surfactant on the polymerization time of the formed nanoparticle was considerably not significant at the concentration range taken. It was inferred that as the temperature of the reaction medium was increased, it resulted in a rapid polymerization rate leading to lesser time taken for the completion of the polymerization rate, where a comparatively quicker precipitation of the nanoparticles was observed. Because at higher temperature, the heat input is rapid thus leading to higher reaction rates and results in polymerization in shorter times.31)
Impact of the Factors on the Response EEThe impacts of the factors on EE are presented in Figs. 3(c) and 3(d). The effect of different parameters on EE were inferred from the DoE studies and from the analysis it was observed that at lower monomer concentration, the EE was found to be less and was increased along with the monomer concentration. This might be attributed to the smaller length of the polymer chain or small amount of the polymer formed resulting in lesser amount of drug encapsulation at lower monomer levels. The observed result was found similar to those reported by Salawi et al.32) It was also observed that as the concentration of the initiator was increased, the entrapment efficiency was found to be decreased. It might be attributed to the rapid polymerization at higher initiator concentrations that might hinder the entry of drug into the polymeric NPs. The effect of surfactant level was found to have positive effect of the EE. It was inferred from the analysis that as the surfactant concentration increases, the entrapment efficiency increases. This could be attributed to the decrease in interfacial tension by the surfactant between the less polar polymer and high polar drug thus increasing the association of the drug within the polymer.33) Theoretically, the increase in the temperature results in increase in the entrapment efficiency of the drug. This might be because the increase in temperature may lead to the increase in the initiator decomposition leading to a formation of large chain length and increase in particle size.34) But, in the 60–70 °C range of the temperature taken in this study, its effect on the EE was found statistically insignificant.
Impact of the Factors on the Response PSThe impacts of the factors on PS are presented in Figs. 4(a) and 4(b). From the DoE analysis, different factors and their impact on the desired response polymerization time were evaluated. The higher the concentrations of the monomer, the particle size of the formed nanoparticles was found to be bigger. This might be due to (i) continued growth of the polymer at higher monomer levels, or (ii) increased diffusion of the monomer from the medium into the developing polymer phase.35) The initiator concentration was increased; a broad particle size distribution was observed which might be attributed to the increase in the growth rate of the oligomeric chains leading to the increased polymer chain lengths.36,37) Theoretically, as the surfactant concentration increases, the particle size decreases. This might be due to the adsorption of the surfactant at the interface between the polymerized nanoparticle and the aqueous reaction medium.38) These adsorbed surfactant molecules develop steric repulsion among the nanoparticles and thus hinder the growth process.39) It was observed that with the increase in the temperature resulted in increase in the particle size which might be attributed to the rapid decomposition of the initiator into free radicals at higher temperatures which might result in the increase in the solvency of the continuous phase (for the developing polymer) thereby the oligomeric chain length might be increased before it got precipitated out.40) Another reason might be that at the higher kinetic energy at higher temperature might cause more collisions of the particles that led to greater final particle size.41) However, it was observed after statistical analysis that in the current research work, the effect of temperature on the particle size of the formed nanoparticles was considerably not significant in the range taken.
Impact of the Factors on the Response kThe impacts of the factors on k are presented in Figs. 4(c) and 4(d). From the DoE analysis studies, it was found that at lower monomer concentration, the dissolution rate was found to be increased. This might be attributed to the lesser amount of polymer coated at low monomer concentration owing to a smaller particle size and greater surface area.35) Whereas at higher monomer concentration, the particle size was more with greater polymerization and thus the drug release might be prolonged. Whereas if the initiator concentration was increased, the dissolution rate was found to be decreased. This might be attributed to the higher number of free radicles formed during decomposition leading to the formation of a longer polymer chain length.36,37) This increased polymer chain length might bind the drug tightly which might cause slower drug release.42,43) Theoretically, as the surfactant concentration increases, the drug release rate also increases owing to the smaller size of the nanoparticle formed due to the absorption of the surfactant to the nanoparticles formed leading to a higher surface area. But in the research work, the effect of surfactant on the drug release rate of the formed nanoparticle was considerably not significant at the concentration range taken. It was inferred that similar to the initiator concentration, as the temperature of the reaction medium was increased, the dissolution rate was found to be decreased. This might be attributed to the more number of free radicles with higher kinetic energy and collisions would result in forming longer polymer chains.41) This might cause tight binding of the drug thus resulting in slower release.
OptimizationThe prime objective of this work was to develop polymeric nanoparticles for a high soluble drug with sufficiently high EE and significant controlled release from monomers by employing polymerization technique in as less time as possible. In this context, the quality target of the LPANs for the formulation optimization was set as minimum PT, maximum EE, minimum PS and minimum k. Graphical optimization by desirability functions approach at this desirability criteria was done and the resultant overlay plot is presented as Fig. 5. Several combinations of the factors on x- and y-axes were tried to represent the design space. Among all the combinations, the Factors A and D when taken on x- and y-axes, showed maximum design space. The design space satisfying the requirement of the desired responses was indicated in the yellow color area. In this design space, the combination of any levels of the factors would yield the LPANs with the desired responses values to the maximum satisfaction. One such combination with maximum desirability was observed at 8.7% (w/v) of the acrylonitrile, 0.4% (w/v) of potassium persulphate, 1.5% (w/v) of SLS and 65 °C of polymerization temperature was identified as the optimized formulation. The predicted values of the responses for this optimized LPANs were given by the software as 96.3 min. of PT, 80.2% of EE, 247 nm of PS and the drug release rate constant of 0.27 h−1. An LPANs formulation at the optimized combination was prepared again and characterized. The results of the responses were observed as 91.7 min of PT, 81.4% of EE, 253 nm of PS and the drug release k value of 0.262 h−1 (18 h to release 99%). These observed values of the responses were found to fit within 95% confidence intervals of those of the predicted values. Hence, these observations designate that the formulation optimization of the LPANs were successfully achieved by this statistical analysis.
DSC StudiesThermal analysis was applied to study the physical state of LMV in the formulation and to check the drug-excipient compatibility. DSC was carried out for the drug and optimized formulation. In the spectrum of pure LMV, the sharp endothermic peak of LMV as shown in Fig. 6(a) was observed at 180.5 °C. This peak is attributed to the melting point of crystalline LMV used in the formulations. Whereas the spectrum of LPAN, as shown in Fig. 6(b), exhibited a similar sharp endotherm at 179.2 °C. As there was no significant change in the peak position in the prepared LPANs against the pure LMV, it could be inferred that either monomer or the developed polymer did not show any incompatibility with the LMV. Further, the LMV remained in the original crystalline form even in the formed LPANs. Further, an additional broad peak in the spectrum of LPANs was observed at 77.2 °C. This peak was corresponding to the polymer PAN as it has a glass transition temperature around 80 °C as reported by many authors.44) Hence, the DSC studies confirm the formation of PAN and also crystallization of the LMV inside the nanoparticles.
TEM AnalysisThe TEM images shown in Fig. 7 illustrated that the LPANs were formed almost spherical in shape during the dispersion polymerization technique. Further, the surfaces of the individual nanoparticles also look uniform and continuous without any surface irregularities. These results inferred that the process conditions and formulation were suitable enough to produce spherical LPANs by polymerization technique.
Cytotoxicity Studies of LPANsThe cell viability assay by MTT assay method was performed to determine the cytotoxicity of the prepared LPANs using the polymerization of the monomers. The results are displayed as the % viability plot against the concentration and shown in Fig. 8. Using the regression equation, the IC-50 (concentration required to kill 50% of the cells)45) was calculated and was found to be 52.14 µg/mL. Considering the volume of distribution of LAM which is around 91 L,46) its maximum dose (300 mg) which was contained in approximately 1125 mg of the optimized LPANs (calculated based on the quantities in optimized formulation and its %EE value) would yield a total maximum plasma concentration of 12.36 µg/mL. This concentration was 4.2 times lesser than the observed IC-50 of the LPANs. These observations clearly signified that the optimized LPANs prepared by dispersion polymerization technique were completely safe to use.
Conclusion
The purpose of this study was to optimize the preparation of PAN nanoparticles by in-situ polymerization with LMV as model drug by using Box Behnken Design. Using dispersion polymerization method, the drug was successfully incorporated into the PAN NPs during in- situ polymerization. Formulation variables including monomer concentration, initiator concentration and surfactant concentration, process variable temperature have shown significant effect on LPANs characteristics such as PT, EE, PS and drug release rate. Optimization of the factors was done so as to achieve the LPANs with high drug entrapment and more controlled release. LMV was compatible and stable without any changes in the formed LPANs as shown by the DSC studies. The LPANs were found safe by the cytotoxicity studies. These results altogether confirmed that ADP could be a simple, economical yet effective approach in developing LPANs with desired characteristics.
Acknowledgments
The authors wish to express their gratitude to the Vignan’s Foundation for Science Technology and Research, Guntur, India and GITAM University, Hyderabad for arranging the necessary facilities in completion of the work.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Englert C., Brendel J. C., Majdanski T. C., Yildirim T., Schubert S., Gottschaldt M., Windhab N., Schubert U. S., Prog. Polym. Sci., 87, 107–164 (2018).
- 2) Sung Y. K., Kim S. W., Biomater. Res., 24, 12 (2020).
- 3) Sivalingan G., Gnk G., Chandrasekaran M., Res. J. Pharm. Technol., 13, 3501–3507 (2020).
- 4) Zielińska A., Carreiró F., Oliveira A. M., Neves A., Pires B., Venkatesh D. N., Durazzo A., Lucarini M., Eder P., Silva A. M., Santini A., Souto E. B., Molecules, 25, 3731 (2020).
- 5) Begines B., Ortiz T., Pérez-Aranda M., Martínez G., Merinero M., Argüelles-Arias F., Alcudia A., Nanomaterials (Basel), 10, 1403 (2020).
- 6) Gagliardi A., Giuliano E., Venkateswararao E., Fresta M., Bulotta S., Awasthi V., Cosco D., Front. Pharmacol., 12, 601626 (2021).
- 7) Reis C. P., Neufeld R. J., Ribeiro A. J., Veiga F., Nanomedicine (Lond.), 2, 8–21 (2006).
- 8) Charleux B., Ganachaud F., “Macromolecular Engineering,” 1st ed., ed. by Matyjaszewski, Krzysztof, Gnanou, Yves, Leibler, Ludwik, Wiley, 2007, pp. 605–642.
- 9) Zheng C., Huang Z., J. Dispers. Sci. Technol., 40, 1317–1325 (2019).
- 10) Hassan Hefni H., Basiouny N., Abouelmagd W., Abd El-Rahman N., Esmail E., Youssif M., Egypt. J. Chem., 65, 1347–1353 (2022).
- 11) Lee J.-M., Kang S.-J., Park S.-J., Macromol. Res., 17, 817–820 (2009).
- 12) Boguslavsky L., Baruch S., Margel S., J. Colloid Interface Sci., 289, 71–85 (2005).
- 13) “Data Sources—PubChem.”: ‹https://pubchem.ncbi.nlm.nih.gov/source/hsdb/2927%20;%20https://www.chembk.com/en/chem/Polyacrylonitrile›, cited 23 May, 2024.
- 14) Sadrearhami Z., Morshed M., Varshosaz J., Fibers Polym., 16, 254–262 (2015).
- 15) Lian Q., Liu H., Zheng X., Jia D., Liu C., Wang D., RSC Adv., 10, 15715–15725 (2020).
- 16) Lv H., Guo S., Zhang G., He W., Wu Y., Yu D.-G., Polymers (Basel), 13, 4286 (2021).
- 17) Waghule T., Dabholkar N., Gorantla S., Rapalli V. K., Saha R. N., Singhvi G., Biomed. Pharmacother., 141, 111940 (2021).
- 18) Sharaf N. S., Shetta A., Elhalawani J. E., Mamdouh W., Polymers (Basel), 14, 144 (2021).
- 19) Czyrski A., Sznura J., Sci. Rep., 9, 19458 (2019).
- 20) Mohy Eldin M. S., Elaassar M. R., Elzatahry A. A., Al-Sabah M. M. B., Arab. J. Chem., 10, 1153–1166 (2017).
- 21) Ogunwuyi O., Kumari N., Smith K. A., Bolshakov O., Adesina S., Gugssa A., Anderson W. A., Nekhai S., Akala E. O., Infect. Dis. (Auckl.), 9, 21–32 (2016).
- 22) Adesina S. K., Wight S. A., Akala E. O., Drug Dev. Ind. Pharm., 40, 1547–1556 (2014).
- 23) Zhao Z. M., Wang Y., Han J., Zhu H. D., An L., Chem. Pharm. Bull., 63, 180–186 (2015).
- 24) Kimura K., Yamasaki K., Nakamura H., Harakate M., Taguchi K., Otagiri M., Chem. Pharm. Bull., 66, 382–390 (2018).
- 25) Yu M., Yuan W., Li D., Schwendeman A., Schwendeman S. P., J. Control. Release, 315, 23–30 (2019).
- 26) Kim D. M., Lee G. D., Aum S. H., Kim H. J., Biol. Pharm. Bull., 31, 1704–1710 (2008).
- 27) Mehrotra A., Nagarwal R. C., Pandit J. K., Chem. Pharm. Bull., 59, 315–320 (2011).
- 28) Dauletov Y., Abdiyev K., Toktarbay Z., Nuraje N., Zhursumbaeva M., Kenzhaliyev B., Fibers Polym., 19, 2023–2029 (2018).
- 29) Kongkaew A., Wootthikanokkhan J., J. Appl. Polym. Sci., 75, 938–944 (2000).
- 30) Przesławski G., Szcześniak K., Gajewski P., Marcinkowska A., Polymers (Basel), 14, 5005 (2022).
- 31) Li M.-X., Lee D., Lee G. H., Kim S. M., Ben G., Lee W. I., Choi S. W., Polymers (Basel), 12, 1133 (2020).
- 32) Salawi A., Khan A., Zaman M., Riaz T., Ihsan H., Butt M. H., Aman W., Khan R., Majeed I., Almoshari Y., Alshamrani M., Polymers (Basel), 14, 2369 (2022).
- 33) Pandita D., Ahuja A., Velpandian T., Lather V., Dutta T., Khar R. K., Pharmazie, 64, 301–310 (2009).
- 34) Chaudhari K. R., Ukawala M., Manjappa A. S., Kumar A., Mundada P. K., Mishra A. K., Mathur R., Mönkkönen J., Murthy R. S. R., Pharm. Res., 29, 53–68 (2012).
- 35) Lok K. P., Ober C. K., Can. J. Chem., 63, 209–216 (1985).
- 36) Liu B., Wang Y., Zhang M., Zhang H., Polymers (Basel), 8, 55 (2016).
- 37) Carro S., Herrera-Ordonez J., Castillo-Tejas J., J. Polym. Sci. A Polym. Chem., 48, 3152–3160 (2010).
- 38) Sarheed O., Dibi M., Ramesh K. V. R. N. S., Pharmaceutics, 12, 1223 (2020).
- 39) Dong Y., Chang Y., Wang Q., Tong J., Zhou J., Bull. Mater. Sci., 39, 35–39 (2016).
- 40) Omirova R. Zh., Bolysbek A. A., Shirinov Sh. D., Dzhalilov A. T., Rasayan J. Chem., 12, 2047–2051 (2019).
- 41) Bai X., Wang X., Pu X., e-Polymers (Basel), 13, 1–9 (2013).
- 42) Khattabi A. M., Talib W. H., Alqdeimat D. A., Saudi Pharm. J., 26, 1022–1026 (2018).
- 43) You X., Wang L., Zhang J., Tong T., Dai C., Chen C., Wu J., Chin. Chem. Lett., 34, 107720 (2023).
- 44) Burkanudeen A., Krishnan G. S., Murali N., J. Therm. Anal. Calorim., 112, 1261–1268 (2013).
- 45) Hazekawa M., Nishinakagawa T., Kawakubo-Yasukochi T., Nakashima M., Exp. Ther. Med., 18, 3197–3205 (2019).
- 46) Johnson M. A., Moore K. H. P., Yuen G. J., Bye A., Pakes G. E., Clin. Pharmacokinet., 36, 41–66 (1999).