Regular Article
Synthetic Study towards Providencin: Stereocontrolled Synthesis of the Furan-Substituted Cyclobutanol Segment
2024 Volume 72 Issue 11 Pages 966-969
Details
2024 Volume 72 Issue 11 Pages 966-969
This study explores the synthesis of unique furanocembranoid-type marine diterpenoid, providencin. Providencin features a highly oxidized structure with two furan rings, two oxirane rings, and a bicyclo[12.2.0]hexadecane framework. Its potential as a lead compound for drug development has drawn attention to its total synthesis, particularly focusing on the challenging right-half segment involving a highly substituted cyclobutane ring. We developed a novel synthetic strategy for the fragment using a [2 + 2] cycloaddition reaction of lithium ynolates with α,β-unsaturated lactone, successfully constructing a bicyclic cyclobutene structure. Stereoselective hydrogenation of cyclobutenes was achieved by using Crabtree’s catalyst under high pressure H2 atmosphere. After further transformation, the synthesis of the furan-substituted cyclobutanol fragment having a formyl side chain was accomplished.
Natural organisms, such as plants and fungi, have acquired the ability to synthesize various secondary metabolites during the course of biological evolution, leading to the discovery of natural products with diverse molecular structures. For example, various terpenes are synthesized starting from isoprene, through the biosynthesis of linear molecules such as geraniol and farnesol. These molecules undergo cyclization and rearrangement reactions under the action of specific enzymes, forming structurally unique cyclic structures, which are further synthesized through redox reactions. As a result, terpenoids with structures previously unimaginable in traditional synthetic chemistry based on reaction development are often discovered, attracting attention in drug discovery research and functional molecule development. Providencin (1), shown in Fig. 1, is a furanocembranoid-type diterpenoid1–4) isolated and structurally elucidated by Marrero et al. in 2003 from the Caribbean gorgonian octocoral, Pseudopterogorgia kallos.5) The unique structure of 1 is characterized by a highly oxidized structure containing a furan ring, a butanolide ring and two oxirane rings, with a bicyclo[12.2.0]hexadecane framework. Providencin exhibits moderate in vitro cytotoxicity against human breast (MCF7), lung (NCI-H-460), and CNS (SF-286) cancer cell lines. Due to its unique structure and potential as a lead compound in pharmaceuticals, providencin has become an attractive target for total synthesis among many synthetic chemists. However, total synthesis of 1 has not yet been achieved, although semi-synthesis from biosynthetically related natural product was reported.6)

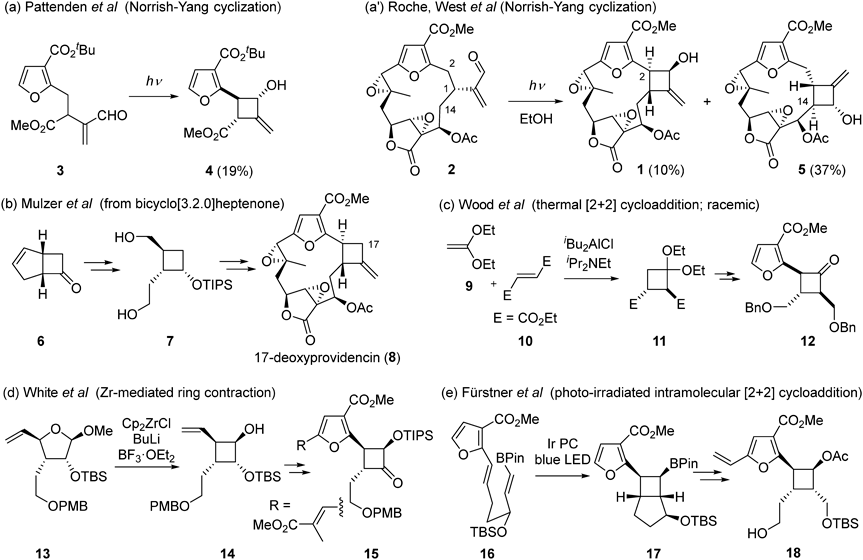
Chart 1 summarizes the synthetic studies of synthetically challenging right-half segment of 1, specifically the highly substituted cyclobutane ring. Bray and Pattenden proposed that the cyclobutane ring may be formed through the Norrish-Yang cyclization reaction from bipinnatin E (2) leading to the biosynthesis of providencin.7) They successfully constructed the model cyclobutane 4 from enal 3 despite in low yield (Chart 1a). Recently, Roche and West achieved semi-synthesis of providencin (1) from bipinnatin E (2) and proved the biosynthetic pathway,8) however, in the transformation, regioisomeric cyclobutane 5, cyclized at the C(14) position, was obtained as the major product6) (Chart 1a′). On the other hand, many researchers have attempted to synthesize the cyclobutane ring without relying on the biosynthetic route. As a pioneering and most advanced work, Mulzer and colleagues reported the synthesis of 17-deoxyprovidencin (8).9–12) The highly substituted four-membered ring was synthesized by ozonolysis-reduction of optically active bicyclo[3.2.0]heptenone (6), followed by construction of the furan ring to obtain 8 via tri-substituted cyclobutane 7 (Chart 1b). However, they did not succeed in a late-stage oxidation at the C(17) position of 8. Wood’s group synthesized a highly substituted cyclobutane 11 by a thermal [2 + 2] cycloaddition reaction of diethylketene acetal (9) and diethyl fumarate (10).13) Then, nucleophilic addition–rearrangement sequence to introduce the furan ring afforded tri-substituted cyclobutanone 12 in a diastereoselective manner (Chart 1c). White and Jana achieved a ring-contraction reaction of 13, derived from D-glucose in several steps, by zirconocene(0) reagent to give tetra-substituted cyclobutane 14 stereoselectively, followed by further steps to synthesize 1514,15) (Chart 1d). Recently, Spohr and Fürstner reported the synthesis of the right-half segment 18 using an intramolecular [2 + 2] cycloaddition reaction of diene 16 under LED irradiation to synthesize bicyclic compound 17.16) However, challenges remain in controlling the diastereoselectivity of the [2 + 2] cycloaddition (Chart 1e).
We recently developed a [2 + 2] cycloaddition reaction of highly reactive lithium ynolates with α,β-unsaturated carbonyl compounds to give highly substituted cyclobutenes.17) We envisaged that [2 + 2] cycloaddition reaction of ynolate 19 with 5,6-dihydro-2H-pyran-2-one (20) followed by acylation of enolate intermediate 21 would afford bicyclic cyclobutene 22. Further transformation of 22 may allow stereocontrolled synthesis of tetracyclic cyclobutane 23 (Chart 2). In this paper, we report a new stereoselective entry to construct the right-half segment of providencin (1).

Tribromomethyl ketone precursors 26a and 26b for the synthesis of ynolates were prepared in four steps from 3-furanmethanol (24) (Chart 3). After directing group assisted deprotonation of 24 followed by formylation, the hydroxy group of the corresponding product was protected with bulky silyl groups, triisopropylsilyl (TIPS) and t-butyldiphenylsilyl (TBDPS), to afford 25a and 25b, respectively. Then, tribromomethyl anion was added to 25a and 25b, followed by oxidation of the secondary hydroxy group to obtain 26a and 26b, respectively. In accordance with our reported procedure,17) the [2 + 2] cycloaddition reaction of lithium ynolates 19, prepared from 26, with 5,6-dihydro-2H-pyran-2-one (20), followed by trapping of the resulting lithium enolate 21 with acyl anhydride, gave bicyclic cyclobutenes 22 in moderate yield (see also, Chart 2). Due to the instability and high reactivity of the reaction intermediate ynolate 19 and enolate 21, careful control of temperature and reaction time was required at each stage to obtain the desired product 22.

Although various attempts for hydrogenation of bicyclic cyclobutene 22a were investigated, no formation of the desired cyclobutane and the recovery of the starting material 22a were observed. Next, we decided to investigate hydrogenation after conversion to monocyclic cyclobutene 27. Transesterification of 22a under weak basic conditions, followed by t-butyldimethylsilyl (TBDMS) protection of a generated hydroxy group, afforded cyclobutene 27a in low yield. Two side reactions were observed as reasons for the low production of 27a: the first is the reverse lactonization of the resulting ring-opened hydroxyester into 22a during purification and silylation reaction; the second is the decomposition of the enol acetate moiety during transesterification reaction. Despite of the low yield of the monocyclic cyclobutene, we continued to examine stereoselective reduction of 27a using Crabtree’s catalyst, [Ir(Py)(PCy)3(COD)]PF6,17–19) in which the ester group would work as a directing group to coordinate iridium atom. The desired reaction did not proceed under H2 gas at atmospheric pressure (see, Supplementary Materials), but slow reduction occurred under high pressure conditions (15 atm, H2) using an autoclave. However, under the conditions, the two silyl groups of 27a were deprotected by proton in situ generated from the catalyst and H2,20,21) followed by spontaneous lactonization to form bicyclic cyclobutane 28 with a trace amount, whose production was proved by MS; 322) (Chart 4). The monocyclic cyclobutane 23 was not observed under these conditions.

To suppress undesired lactonization, the TBDMS group of 27 was changed into acetyl groups. The reaction of 22a in MeOH in the presence of catalytic amounts of p-TsOH resulted in transesterification and unexpected desilylation to give the ring-opened dihydroxyester, which was acetylated to 27c (2 step yield of 56%). During isolation of the diol intermediate, re-lactonization still occurred, resulting in a moderate yield. Hydrogenation of 27c with Crabtree’s catalyst under 15 atm hydrogen atmosphere stereoselectively gave 23c in 60% yield as a single diastereomer (Chart 5). The stereochemistry of 23c was determined by nuclear Overhauser effect (NOE) experiment.

Since the TIPS group in the furan side chain was cleaved during the transesterification reaction of 22a under acidic conditions, the protecting group was changed to the more resistant TBDPS group (22b). The ring-opening reaction, followed by acetylation, afforded 27b albeit low yield, and it was proved that TBDPS group was retained during the conversion. The stereoselective reduction was then carried out according to the same conditions, resulting in the conversion to 23b. The acetyl group of 23b was selectively deprotected by transesterification, and the resulting alcohol was oxidized by IBX to give aldehyde 29 in 43% (2 steps) (Chart 6). The formyl group of 29 can be used as a point to construct macrocyclic ring of providencin (1) at the C(13) position.12,15)

In this study, we focused on the right-half cyclobutane segment, of providencin (1), a compound with a unique structure that serves as an attractive target for synthetic chemists as a potential lead compound for drug development. We proposed a novel synthetic strategy using [2 + 2] cycloaddition reaction of lithium ynolate and α,β-unsaturated carbonyl compounds, successfully constructing a bicyclic cyclobutene structure. We achieved stereoselective hydrogenation of cyclobutenes by using Crabtree’s catalyst under high pressure to give tetra-substituted cyclobutanes. However, challenges remain in optimizing the yields at each stage, in the selection of appropriate protecting groups and in the asymmetric synthesis, indicating the need for further research.
This work was supported by JSPS KAKENHI (Grant Numbers: 21K05069, 23K18183, 23K27295, and 24K09721) and MEXT KAKENHI (Grant Number: JP21H05211) in Digi-TOS, BINDS from AMED (Grant Numbers: 22ama121042j0001 and 22ama121034j0001), Takeda Foundation and Kobayashi Foundation.
The authors declare no conflict of interest.
This article contains supplementary materials.