Abstract
A total synthesis of javaberine A was achieved through a lithium amide-mediated intramolecular hydroamination of an N-allyl aminoalkene. The desired hydroamination was accomplished using an excess of i-Pr2NH with a substoichiometric amount of n-BuLi. Using an excess of both n-BuLi and i-Pr2NH led to tandem cyclization, however, resulting in the construction of a tricyclic structure through the formation of one C–N and two C–C bonds in a single operation. Additionally, epimerization of the H8-H14 cis-benzyl tetrahydroisoquinoline to the trans isomer was achieved via β-elimination followed by intramolecular hydroamination.
Introduction
Javaberine A (1), a class of berberine alkaloids, inhibits lipopolysaccharide-induced tumor necrosis factor-α and nitric oxide production.1) Related berberine alkaloids with C8-substituents have attracted increased attention due to their intriguing biological activities. For example, theoneberine (2) exhibits antimicrobial activity against Gram-positive bacteria,2) and O-methylcorytenchirine (3)3) and coralydine (4)4) inhibit human CYP5) (Fig. 1). The tetrahydroprotoberberine core structure contains two tetrahydroisoquinoline moieties. The total synthesis of these alkaloids typically involves isoquinoline synthesis methods, such as Bischler–Napieralski cyclization,6) Pictet-Spengler cyclization,7,8) intramolecular N-alkylation,6) intramolecular hydroamination,7) and others.9–12) Alternative synthetic approaches include Stevens rearrangement for the installation of C8-substituents.13) The relative stereochemistry between H8 and H14 is trans in javaberine A (1), theoneberine (2), and O-methylcorytenchirine (3), whereas a cis relationship is observed in coralydine (4). In this context, we studied the enantio- and diastereoselective synthesis of C8-substituted berberines via Bischler–Napieralski cyclization followed by reduction or alkylation of the iminium intermediate.14–17) We recently reported the synthesis of the javaberine A framework trans-7 through sequential hydroamination of dienylamine 518) (Chart 1). Although this methodology provided the framework for javaberine A in a trans-selective manner, the chemical yield was limited due to an undesired elimination reaction of 6-Li, generated from 6 with n-BuLi, producing dihydroisoquinoline 8 and styrene derivative 9. Protecting the amino group of dienylamine 5 during the initial cyclization would effectively suppress this side reaction and allow intramolecular hydroamination of the secondary amine X to afford the corresponding cyclized product. Subsequent N-deprotection would yield isoquinoline 6. Successive cyclization18) and O-demethylation14) led to javaberine A (1), as previously reported. Herein, we report the racemic total synthesis of javaberine A (1) based on a stepwise hydroamination strategy. During this study, we encountered an unexpected tandem ring-forming reaction. Additionally, we developed the transformation of H8-H14 cis tetrahydroprotoberberine cis-7 to trans-7 by eliminating cis-7 to form 6, followed by a re-cyclization process.
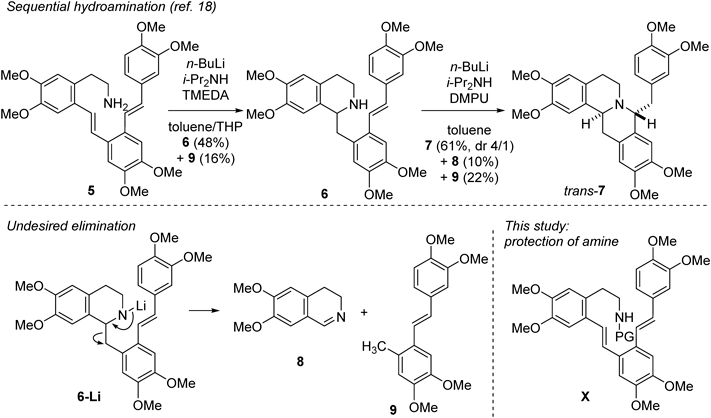
Chart 1. Sequential Hydroamination of Dienylamine 5 and Undesired Elimination Reaction
Results and Discussion
Considering the nucleophilicity of secondary amine X and the ease of deprotection, we selected N-allylamine 14 as the cyclization substrate. Dienylamine 5 was synthesized from commercially available 3,4-dimethoxystyrene 10 and 2-bromo-3,4-dimethoxybenzaldehyde 11 in five steps, as previously reported.18) N-tert-Butoxycarbonyl (Boc) protection of 5 (87%) followed by allylation (95%) and subsequent N-Boc deprotection (81%) provided the cyclization precursor of N-allylamine 14 (Chart 2).

Chart 2. Synthesis of Cyclization Precursor 14
With the cyclization precursor in hand, we next examined lithium amide-mediated hydroamination of N-allylamine 14 using 5 equivalents (equiv.) of n-BuLi and i-Pr2NH. Contrary to our expectations, however, the desired cyclized product 15 was not obtained. Instead, unexpected tandem cyclized product 16 was obtained in 40% yield (Table 1, entry 1). When excess i-Pr2NH was used with respect to n-BuLi, the desired product 15 was obtained (entries 2, 3). Adding n-BuLi (1 equiv.) to the solution of 14 and i-Pr2NH (10 equiv.) in tetrahydrofuran (THF) at −40 °C with stirring at 0 °C for 23 h afforded the desired cyclized product 15 in 25% yield, along with 16 in 49% yield (entry 2). The use of a substoichiometric amount of n-BuLi (0.4 equiv.) suppressed the formation of the tandem cyclized product 16, and the desired monocyclized product 15 was obtained in 49% yield (entry 3).19)
Table 1. Hydroamination of
N-Allylamine
14 |
|---|
| Entry | n-BuLi (equiv.) | i-Pr2NH (equiv.) | Temp, time | 15 (%) | 16 (%) |
|---|
| 1 | 5 | 5 | r.t., 2 h | 0 | 40 |
| 2 | 1 | 10 | −40 to 0 °C, 15 min, then 0 °C, 23 h | 25 | 49 |
| 3 | 0.4 | 2 | −60 to 0 °C, 1.5 h, then 0 °C, 15 h | 49 | 0 |
A plausible reaction pathway is shown in Chart 3. Deprotonation of N-allylamine 14 by n-BuLi and/or lithium diisopropylamide produces the corresponding lithium amide 14-Li. Subsequent intramolecular aminolithiation of the styrene moiety leads to cyclization to give 15-Li. The desired pathway involves protonation of 15-Li by diisopropylamine, yielding cyclized product 15. Alternatively, tandem cyclization of intermediate 15-Li can occur; 5-exo cyclization to the olefin of the allyl group forms alkyllithium 17, followed by 6-exo cyclization to the styrene moiety and subsequent protonation, resulting in tandem cyclized product 16. The use of excess i-Pr2NH with respect to n-BuLi favors the protonation of 15-Li, leading to the desired product 15 (Table 1, entries 2, 3). On the other hand, using excess n-BuLi promotes tandem cyclization (Table 1, entry 1). This tendency is consistent with our previous report on aminolithiation–carbolithiation consecutive cyclization of N-allylaminoalkene.20,21) Although the formation of 16 was undesired for the total synthesis of javaberine A, this tandem cyclization allows for the construction of a tricyclic structure through the formation of one C–N and two C–C bonds in a single operation.

Chart 3. Plausible Reaction Pathway
Tandem cyclization product 16 was obtained as a single diastereomer. The relative stereochemistry of 16 was determined by 1H-nuclear Overhauser effect spectroscopy (NOESY) experiments, as shown in Chart 4. Based on Cohen’s report22) and our previous study,20,21) the aminolithiation of 14-Li, generated from 14 by lithiation, should proceed via a parallel arrangement of N–Li and C=C, oriented toward an equatorial position, to yield 15-Li. Subsequent carbolithiation during 5-exo cyclization, in which the conformation of C–Li and C=C assumes a pseudoequatorial arrangement, produces 17. The second carbolithiation of 6-exo cyclization would also proceed via an equatorial arrangement of the C–Li and C=C bond, yielding 16. Indeed, the relative configuration of Ha, Hb, and Hc in 16 (Chart 4) matched those of the tandem cyclized products obtained through aminolithiation–carbolithiation in our previous study.20,21)

Chart 4. Relative Stereochemistry of 16 and Plausible Stereochemical Pathway
For the total synthesis of javaberine A, we next investigated the N-deallylation of 15. Treatment of 15 with RhCl(PPh3)3 in an acetonitrile–water mixture under reflux conditions afforded N-deallylated product 6 in 84% yield (Chart 5). As previously reported, hydroamination of 618) and the following O-demethylation14) afforded javaberine A. In our recent work, direct hydroamination of primary amine 5 provided cyclized product 6 in 48% yield18) (Chart 1). In this study, we aimed to improve the chemical yield of 6 by protecting primary amine 5 with an allyl group (3 steps, 73% overall yield, Chart 2), followed by hydroamination of N-allylamine 14 (49%, Table 1, entry 3) and N-deallylation (84%, Chart 5). Unfortunately, the overall yield was not improved using this method.

Chart 5. N-Deallylation of 15
We also examined the epimerization of cis-7, corresponding to the epimer of javaberine A, to trans-7. Because both H8-H14 cis and trans alkaloids are present in natural products of C8-substituted tetrahydroprotoberberines,1–4,23) stereoselective construction of C8-substituents is crucial. In this context, we developed a stereoselective synthesis of C8-methyl and benzyl tetrahydroprotoberberines, yielding cis14,16) and trans15,16) derivatives. Notably, cis-7 could be easily synthesized on a gram scale without column chromatography.14) Therefore, the epimerization of cis-7 to trans-7 is a valuable method and could also be useful for structural modification of natural products.
Treatment of cis-7 with 1-chloroethyl chloroformate in dichloroethane, followed by refluxing in methanol, afforded 6 in 61% yield (88% based on the recovery of cis-7) through a process involving N-acylation, β-elimination, and N-deacylation17) (Chart 6). The lithium amide-mediated intramolecular hydroamination of 6 gave trans-7 with moderate trans-selectivity.18) Thus, the epimerization of cis-7 to trans-7 was achieved through this elimination-hydroamination process.

Chart 6. β-Elimination of cis-7 to Aminoalkene 6
Conclusion
We developed a lithium amide-mediated intramolecular hydroamination of N-allylamine 14. The reaction pathway was controlled by balancing the equivalents of n-BuLi and i-Pr2NH; using excess i-Pr2NH with a substoichiometric amount of n-BuLi facilitated the hydroamination, yielding compound 15. In contrast, an excess of both n-BuLi and i-Pr2NH led to tandem cyclization, resulting in the tricyclic structure of compound 16. Additionally, epimerization of the H8-H14 cis-benzyl tetrahydroisoquinoline (cis-7 to trans-7) was achieved via β-elimination followed by intramolecular hydroamination.
Experimental
General Experimental Procedures1H-NMR (500 MHz) and 13C-NMR (125 MHz) were measured in CDCl3 unless otherwise mentioned. Chemical shift values were expressed in ppm relative to an internal reference of tetramethylsilane (0 ppm) in 1H-NMR and CDCl3 (77.0 ppm) in 13C-NMR. The multiplicity of the 13C peaks was assigned based on distortionless enhancement by polarization transfer (DEPT) data. IR spectroscopy of oil samples was measured as neat liquid films, while solid samples were measured using KBr pellets. Column chromatography was performed using silica gel as a stationary phase.
(E)-2-Allyl-1-(2-(3,4-dimethoxystyryl)-4,5-dimethoxybenzyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (15)Under Ar atmosphere, n-BuLi (0.24 mL, 0.38 mmol) was added to a solution of 14 (522 mg, 0.96 mmol) and i-Pr2NH (0.27 mL, 1.91 mmol) in THF (48 mL)24) at −60 °C. The mixture was warmed to 0 °C over 1.5 h and stirred for 15 h. The reaction was quenched with H2O (20 mL), and the mixture was extracted with CH2Cl2 (30 + 20 + 20 mL). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, and concentrated. Column chromatography (50 g, acetone/hexane 1/2) yielded 15 (254 mg, 49%) as a white solid of mp 151–152 °C.
1H-NMR: 2.55 (1H, dd, J = 2.8, 16.2 Hz), 2.87–2.99 (3H, m), 3.21–3.37 (4H, m), 3.44 (3H, s), 3.74 (3H, s), 3.79 (3H, s), 3.82 (1H, m), 3.90 (3H, s), 3.92 (3H, s), 3.93 (3H, s), 5.06 (1H, dd, J = 1.5, 10.1 Hz), 5.16 (1H, dd, J = 1.5, 17.1 Hz), 5.74 (1H, s), 5.90 (1H, m), 6.50 (2H, s), 6.69 (1H, d, J = 15.9 Hz), 6.83 (1H, d, J = 8.3 Hz), 6.92 (1H, d, J = 15.9 Hz), 6.94 (1H, d, J = 2.2 Hz), 6.96 (1H, dd, J = 2.2, 8.3 Hz), 7.06 (1H, s). 13C-NMR: 24.7 (CH2), 38.3 (CH2), 43.5 (CH2), 55.4 (CH3), 55.7 (CH3), 55.8 (CH3), 55.9 (CH3 × 2), 56.0 (CH3), 57.1 (CH2), 61.7 (CH), 108.5 (CH), 109.0 (CH), 111.2 (CH), 111.3 (CH), 111.7 (CH), 114.5 (CH), 117.2 (CH2), 119.5 (CH), 124.5 (CH), 125.8 (C), 128.0 (CH), 128.6 (C), 129.7 (C), 130.4 (C), 131.1 (C), 136.4 (CH), 146.2 (C), 147.5 (C), 147.7 (C), 148.3 (C), 148.7 (C), 149.1 (C). IR (neat): 3024, 2954, 2839, 1605, 1512. High resolution (HR)MS-FAB m/z: [M + H]+ Calcd for C33H40NO6, 546.2856; Found, 546.2861.
Acknowledgments
This work was partially supported by JSPS KAKENHI Grant Number: 24K09718.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
References and Notes
- 1) Shimoda H., Nishida N., Ninomiya K., Matsuda H., Yoshikawa M., Heterocycles, 55, 2043–2050 (2001).
- 2) Kobayashi J., Kondo K., Shigemori H., Ishibashi M., Sasaki T., Mikami Y., J. Org. Chem., 57, 6680–6682 (1992).
- 3) Lu S., Su T., Kametani T., Ujiie A., Ihara M., Fukumoto K., J. Chem. Soc., Perkin Trans. 1, 1976, 63–68 (1976).
- 4) Bruderer H., Metzger J., Brossi A., Daly J. J., Helv. Chim. Acta, 59, 2793–2807 (1976).
- 5) Salminen K. A., Meyer A. L., Jerabkova A., Korhonen L. E., Rahnasto M., Juvonen R. O., Imming P., Raunio H., Phytomedicine, 18, 533–538 (2011).
- 6) Sotomayor N., Domínguez E., Lete E., J. Org. Chem., 61, 4062–4072 (1996).
- 7) Pouihes A., Baltaze P. J., Kouklovsky C., Synlett, 24, 1805–1808 (2013).
- 8) Kayhan J., Wanner M. J., Ingemann S., van Maarseveen J. H., Hiemstra H., Eur. J. Org. Chem., 2016, 3705–3708 (2016).
- 9) Lenz G. R., Yang N. C., Chem. Commun., 1967, 1136–1137 (1967).
- 10) Orito K., Miyazawa M., Kanbayashi R., Tatsuzawa T., Tokuda M., Suginome H., J. Org. Chem., 65, 7495–7500 (2000).
- 11) Beckwith A. L. J., Mayadunne R. T. A., ARKIVOC, 2004, 80–93 (2004).
- 12) Chaumontet M., Piccardo R., Baudoin O., Angew. Chem. Int. Ed., 48, 179–182 (2009).
- 13) Valpuesta M., Diaz A., Suau R., Torres G., Eur. J. Org. Chem., 2004, 4313–4318 (2004).
- 14) Yamamoto Y., Tabuchi Y., Baba A., Hideshima K., Nakano M., Miyawaki A., Tomioka K., Heterocycles, 88, 1311–1321 (2014).
- 15) Kakigi R., Nakano M., Ueno A., Miyawaki A., Tomioka K., Yamamoto Y., Heterocycles, 101, 512–523 (2020).
- 16) Kirii M., Matsuoka J., Miyawaki A., Tomioka K., Yamamoto Y., Heterocycles, 103, 817–826 (2021).
- 17) Uenishi S., Kakigi R., Hideshima K., Miyawaki A., Matsuoka J., Ogata T., Tomioka K., Yamamoto Y., Tetrahedron, 90, 132165 (2021).
- 18) Yamamoto Y., Baba H., Toriyama M., Matsuoka J., Miyawaki A., Tomioka K., Tetrahedron, 151, 133788 (2024).
- 19) In entries 1–3 of Table 1, N-allylamine 14 and allylic protons around 5 ppm were not detected in crude 1H-NMR. The reaction was relatively messy, and unidentified byproducts were produced.
- 20) Tsuchida S., Kaneshige A., Ogata T., Baba H., Yamamoto Y., Tomioka K., Org. Lett., 10, 3635–3638 (2008).
- 21) Yamamoto Y., Yamaguchi T., Kaneshige A., Hashimoto A., Kaibe S., Miyawaki A., Yamada K., Tomioka K., Synlett, 28, 2913–2917 (2017).
- 22) Liu H., Deng K., Cohen T., Jordan K. D., Org. Lett., 9, 1911–1914 (2007).
- 23) Lopes L. M. X., Phytochemistry, 31, 4005–4009 (1992).
- 24) The solubility of the substrate N-allylamine 14 in THF was low, necessitating a large amount of THF to dissolve 14.
